

In this issue of Blood, Dulmovits et al1 show that the high mobility group box-1 protein (HMGB1), which is secreted during sepsis resulting in anemia of chronic disease, blocks red blood cell production by interfering with the binding of erythropoietin (EPO; the hormone that controls red blood cell production) to its receptor (EPOR). Anemia of chronic disease, now more accurately referred to as anemia of inflammation (AI), is second only to iron deficiency as the most common type of anemia. The symptoms of AI include fatigue, sweating, and headaches. Laboratory findings include low reticulocyte counts, high levels of circulating EPO, and low serum iron levels. Although AI is relatively common, it has proven complicated to treat because it does not respond to treatment with EPO, unlike many other forms of anemia.2
The normal regulation of EPO regulation of red blood cell production (erythropoiesis) is well understood. EPO binds to EPOR, stimulating receptor dimerization and phosphorylation of the EPOR cytoplasmic domain. Phosphorylated EPOR then activates signal transducer and activator of transcription (STAT-1/3/5), leading to erythroid cell survival, growth, and differentiation.3
HMGB1 and its function are less well understood. HMGB1 is a nuclear protein released from dying cells or secreted by activated innate immune cells, both leading to inflammation. HMGB1 interacts with several surface receptors including Toll-like receptors and the receptor for advanced glycation end products (RAGE).4 This group had previously used a murine sepsis model in which the gut is ligated and punctured to induce sepsis and anemia. The levels of the usual markers of inflammation, tumor necrosis factor and interleukin-6, were rapidly elevated, but returned to baseline levels in 7 days. The anemia, on the other hand, continued for 30 days and was associated with persistently elevated levels of HMGB1. They further showed that injection of recombinant HMGB1 was sufficient to induce the anemia, firmly establishing that HMGB1 played a significant role in AI.5
But how does HMGB1 thwart the effects of EPO? The simplest hypothesis would be that HMGB1 binds to its receptor RAGE, inhibiting erythroid differentiation. In the Dulmovits et al paper, the authors knocked down RAGE and did observe a decreased level of erythropoiesis in a variety of culture models. When HMGB1 was added to RAGE-depleted cells, the inhibition of erythropoiesis was even greater. But if the receptor for HMGB1 (RAGE) is already reduced, how can HMGB1 and RAGE depletion make things worse? (I was expecting to see rescue.) Clearly, HMGB1 acts on erythropoiesis outside of its interactions with RAGE. The authors then showed that even in the presence of pharmacological levels of EPO, the levels of phosphorylated EPOR and activated STAT5 were reduced in the presence of HMGB1, demonstrating a heretofore unknown interaction of HMGB1 with the EPO signaling pathway.
The authors tested a variety of hypotheses about where HMGB1 might be affecting the EPO signaling cascade and clearly show that HMGB1 does not act downstream of EPOR. They then hypothesized that HMGB1 interfered with the initial step of EPO binding to EPOR. Surprisingly (at least to me), the authors were able to show that HMGB1 indeed did bind to EPOR, although with less affinity than EPO. In a series of clever experiments, the authors show that sequential treatment of erythroid cells with first HMGB1 followed by EPO resulted in a severe inhibition of EPO signaling. In additional experiments, the authors demonstrate that the relative concentrations of HMGB1 and EPO are critical and that high levels of HMGB1 can inhibit the even effects of pharmacological levels of EPO. Finally, the authors used their murine sepsis model to show that treatment with neutralizing antibodies against HMGB1 could restore EPO signaling in vivo.
This paper is paradigm shifting because it shows for the first time that a protein outside of the EPO signaling cascade can regulate erythropoiesis (see figure). This was the result of outside-the-box thinking as well as careful testing of alternative hypotheses. Critical readers will appreciate how carefully and thoroughly the authors considered every alternative hypothesis with critical and well-controlled experiments. It will be interesting to see what genes are regulated through the RAGE signaling pathway. Clinically, it would seem that targeting HMGB1 would be an effective means of stimulating erythropoiesis in at least some AI patients. The studies by Dulmovits et al are a great start in that direction.
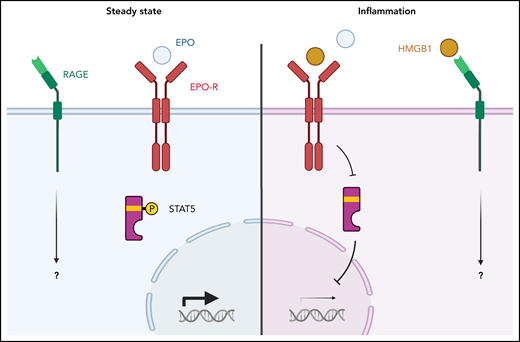
EPO and HMGB1 signaling in erythroblasts. (Left) The normal condition. EPO interacts with its receptor and transduces a signal to STAT5, which activates erythroid genes. (Right) A model of anemia of inflammation. HMGB1 competes with EPO for binding to the EPO receptor. This reduces STAT5 phosphorylation which reduces expression of erythroid genes. An open question is which erythroid genes are regulated by the HMGB1/RAGE signaling pathway.
EPO and HMGB1 signaling in erythroblasts. (Left) The normal condition. EPO interacts with its receptor and transduces a signal to STAT5, which activates erythroid genes. (Right) A model of anemia of inflammation. HMGB1 competes with EPO for binding to the EPO receptor. This reduces STAT5 phosphorylation which reduces expression of erythroid genes. An open question is which erythroid genes are regulated by the HMGB1/RAGE signaling pathway.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal