

In this issue of Blood, Cheon et al1 leverage an integrated, comprehensive genomic approach to better define the molecular characteristics of large granular lymphocyte leukemia (LGLL) subtypes. By including the STAT3 mutation status, they defined distinct molecular signatures and STAT3 mutation-specific clinical associations. They also explored the molecular characteristics of these disorders that contribute to lymphoproliferation, such as recurrent mutations in the death domain of FAS and hotspot PIK3R1 mutations. Their finding of epigenetic alterations coupled with STAT mutations offers new clues into the pathogenesis of these disorders.
Mature LGL disorders have recently aroused considerable interest. These diseases are at the intersection of clonal lymphoproliferative diseases, autoimmunity, and chronic inflammation.2 Furthermore, several underrecognized and borderline conditions are likely to be included in this intriguing arena of differential diagnosis (eg, Felty syndrome, hypoplastic myelodysplastic syndromes, and the clonal hematopoiesis of indeterminate potential). Therefore, a better understanding of the biological and clinical features of these overlapping diseases should lead to more accurate diagnoses and better clinical management.
Mature LGL neoplasms are rare disorders encompassing remarkable phenotypic and genotypic heterogeneity. The clinical presentations are similarly divergent, ranging from indolent to aggressive, with different treatments required. The World Health Organization (WHO) classification of LGLL so far includes patients with chronic proliferation of T (T-LGLL), NK (chronic lymphoproliferative disorder of natural killer lymphocytes [CLPD-NK, still provisional]), and aggressive NK leukemia [ANKL]) lymphocytes. However, the landscape is getting busier. Evidence has been accumulating that T-LGLL is actually 2 distinct clinicopathological subtypes, the more common CD8+ T-LGLL and the less frequent CD4+ T-LGLL with CD4 being expressed, either alone or with CD8dim. Likewise T-LGLL can be divided by surface T-cell receptor expression with Tα/β-LGLL and Tγ/δ-LGLL subsets. Different mutations, including signal transducer and activator of transcription-3 (STAT3), STAT5b, and TET2, just to mention the most relevant, can be harbored in leukemic cells in T-LGLL, CLPD-NK, and ANKL, further subdividing mutated from wild-type patients. The upcoming fifth edition of the WHO classification of hematolymphoid neoplasms will have to take these variants into account.
The hypothesis that an antigen-driven, aberrant immune response underlies the pathogenesis of LGLL is tantalizing, but its etiology remains elusive. STAT signaling is central in directing cells toward survival, as STAT is an inducer of transcription of many prosurvival genes. STAT mutations are unlikely to be the inciting trigger, but compelling evidence indicates that they represent an acquired event during the course of disease that confers an advantage on clone development. Supporting the role of this activator pathway is the finding that ∼40% of patients with LGLL3 have mutations in STAT3 and STAT5b. These mutations are the commonest gain-of-function genetic lesions up to now identified in patients with LGLL (see the figure legend for details).
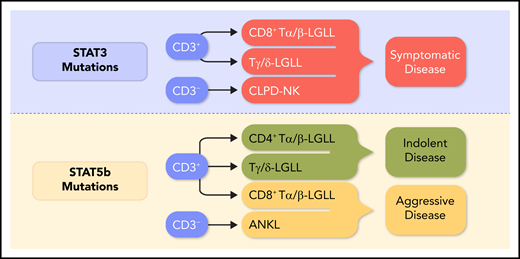
The current status of relationships between STAT mutations and clinical features according to recently published studies.3,5,6,8 Y640F and D661Y are the most frequent STAT3 genetic lesions. For the STAT5b gene, the most common mutations are N642H and Y665F, which are largely found in the SH2 domain. Other less frequent point mutations and insertion or deletions have been found, including the hotspot PIK3R1 mutations and the recurrent mutations in the death domain of FAS, as reported by Cheon et al. The resulting constitutive activation of STAT3 and STAT5b genes translates both into global hypermethylation and into the upregulation of expression of genes that are required for cytokine signaling, cell proliferation, and survival, such as c-Myc, cyclins D1 and D2, Bcl-xl, and Mcl1. Despite the high incidence of STAT genetic lesions, other recurrently mutated genes have been found, including TNFAIP3 and, less frequently, BCL11B, FLT3, and PTPN23 in T-LGLL. Professional illustration by Somersault18:24.
The current status of relationships between STAT mutations and clinical features according to recently published studies.3,5,6,8 Y640F and D661Y are the most frequent STAT3 genetic lesions. For the STAT5b gene, the most common mutations are N642H and Y665F, which are largely found in the SH2 domain. Other less frequent point mutations and insertion or deletions have been found, including the hotspot PIK3R1 mutations and the recurrent mutations in the death domain of FAS, as reported by Cheon et al. The resulting constitutive activation of STAT3 and STAT5b genes translates both into global hypermethylation and into the upregulation of expression of genes that are required for cytokine signaling, cell proliferation, and survival, such as c-Myc, cyclins D1 and D2, Bcl-xl, and Mcl1. Despite the high incidence of STAT genetic lesions, other recurrently mutated genes have been found, including TNFAIP3 and, less frequently, BCL11B, FLT3, and PTPN23 in T-LGLL. Professional illustration by Somersault18:24.
Several recent studies have attempted to unravel the pathogenesis of LGL leukemias and to correlate immunophenotype, genomic analysis, and clinical features.1,3-8 In this regard, the detection of STAT mutations is becoming increasingly important in distinguishing discrete disease subsets.3-5 Symptomatic patients, usually presenting with disease-associated cytopenias, are included in CD8+ Tα/β-LGLL (characterized by a CD3+/CD8+/CD16+/CD56− clone harboring a STAT3 mutation), Tγ/δ-LGLL, and mutated CLPD-NK. Conversely, patients with CD8+ T-LGLL STAT3 wild-type, CD4+ Tα/β-LGLL, and CLPD-NK wild-type are usually asymptomatic (see figure). The role of JAK-STAT alterations has been demonstrated in ANKL (CD2+/CD16+/CD56+), an extremely aggressive NK cell leukemia frequently associated with Epstein-Barr virus.9
The extensive genomic approach used by Cheon et al1 sheds light on unique differences between STAT mutant and wild-type leukemic samples in mutational burden and signatures, transcriptome, and clinical associations. This study and others3-9 have made an important contribution to the LGLL field by expanding our understanding of LGL leukemogenesis and improving classification of these disorders. The use of STAT mutation screening as a diagnostic tool, together with appropriate immunophenotypic analysis, is recommended for accurate characterization of patients with LGLL. As in other hematological conditions, the time has come for molecular genetics in routine diagnostic workup of LGL disorders.
Several questions remain to be addressed. For instance, STAT5b mutation identifies an aggressive disorder that is refractory to therapy when found in CD8 T-LGLL and ANKL, whereas the same mutation in CD4+ T-LGLL does not lead to clinical aggressiveness. Why? One possibility is that the recurrent mutations in chromatin and epigenetic modifying genes, especially KMT2D and SETD1B, that co-occur with STAT3 mutations in LGL leukemia, as discovered by Cheon et al,1 may be the key to interpreting the actual role of STAT in this disorder. As Cheon et al1 mention in the “Discussion” of their article, the use of nonsorted peripheral blood mononuclear cell samples to generate whole exome and RNA-sequencing data represents a potential limitation of their study by not allowing for clear distinction of specific alterations of the leukemic clone from those of the nonleukemic fraction. The latter constraint and the considerable heterogeneity in samples from patients deserve further investigation; to this end, single-cell transcriptomic studies are currently being conducted.10
Beside the molecules that sow the seeds of leukemia, it must be determined how genetic and epigenetic alterations regulate the whole immune cell repertoire that influences immunocompetent cells and their antitumor immune responses, in particular the cross-talk between leukemic cells and nonleukemic mononuclear cells in the microenvironment, including Th17, Treg, and monocytes. These cell populations account for production of interleukin-6 (IL-6), IL-15, CCL5, Fas-L, and other proinflammatory molecules that are central in triggering clonal LGL expansion and its persistence (Cristina Vicenzetto, Vanessa R. Gasparini, Gregorio Barilà, Antonella Teramo, Giulia Calabretto, Samuela Carraro, Valentina Trimarco, Livio Trentin, Monica Facco, Gianpietro Semenzato, and Renato Zambello, manuscript in preparation).
With a growing demand for improved and personalized treatment options in LGL disorders, these studies6-10 should help by providing genetic barcodes to design new compounds (JAK/STAT blockers and demethylating agents, among others) that improve our therapeutic armamentarium for these patients.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal