

In this issue of Blood, describe a new mechanism by which neutrophils adapt to conditions of low oxygen and prolong their lifespan. These findings have important implications for a variety of acute and chronic inflammatory conditions.1
Cells require adenosine triphosphate (ATP) synthesis to support crucial biologic functions during homeostasis and disease. This process becomes impaired during hypoxia.2 Hypoxia is a major stressor involved in various pathologic states, including microbial infections, acute and chronic inflammatory conditions, cancer, trauma, and cardiovascular diseases. Because adapting to hypoxia is key to survival, key adjustments in biologic mechanisms have evolved in mammals. Indeed, oxygen levels in tissue or in nutrient availability are important triggers of metabolic reprogramming. Neutrophils, the first responders against infections and other external insults, must rapidly adapt metabolically to low oxygen levels in tissues to perform their antimicrobial functions. Neutrophils themselves also contribute to decreased oxygen availability at inflamed sites through the generation of reactive oxygen species (ROS). Conventionally, mature neutrophils were considered to have lower mitochondrial mass, and mitochondria were not thought to play major roles in ATP generation in these cells,3 although they did regulate apoptosis3 and neutrophil extracellular trap formation.4 The exact mechanisms by which neutrophils adapt to hypoxia to be able to survive and perform their host defense mechanisms remain incompletely characterized. Willson et al have identified a new role for the mitochondria and the respiratory chain in neutrophil biology by identifying that mitochondrial ROS (mROS) formation stabilizes hypoxia inducible factor- 1α (HIF-1α), a crucial transcriptional regulator that promotes oxygen delivery to hypoxic regions, and that this process relies on the glycerol-3-phosphate (G-3-P) shuttle pathway.
Glucose is a major source of ATP production through glycolysis and the tricarboxylic acid (TCA) cycle. During glycolysis, glucose is converted into pyruvate, and phosphates are transferred to adenosine 5′-diphosphate to generate ATP. Pyruvate can also be converted into acetyl-coenzyme A, which enters the TCA cycle. The TCA cycle produces byproducts that fuel oxidative phosphorylation (OXPHOS) in the mitochondria to produce ATP.2 In addition, the G-3-P shuttle is a pathway that, in certain cells, translocates electrons produced during glycolysis across the inner membrane of mitochondria for OXPHOS, by oxidizing cytoplasmic reduced NAD to NAD+.5 Hypoxia is an important driver of glycolysis, given the limited amount of OXPHOS that is available with low oxygen levels. As glucose becomes crucial in the bioenergetic process during hypoxia, HIF-1 plays a fundamental role, because it induces the expression of glycolytic enzymes that can sustain ATP synthesis under these stress conditions. Indeed, HIF-1 is a crucial transcriptional regulator of inflammatory responses, and its signaling is regulated through the stability of its α subunit. In various cell types, hypoxia can increase mROS, which further affects HIF-1α stability, making the maintenance of mitochondrial membrane potential important in oxygen sensing.2 Stabilization of this molecule has been shown to be important in myeloid function and survival in inflamed tissues exposed to hypoxia.6 Whereas HIF-1α is crucial in establishing proinflammatory effects of hypoxia, dysregulation in its expression can be linked to chronic inflammatory conditions. HIF-1α is expressed in neutrophils, where it may modulate inflammatory responses and environmental adaptation to respond to changes in energy requirements in the context of tissue damage and hypoxia.7 Furthermore, hypoxia seems to have a dual role in neutrophils, because there is evidence that it can be anti-inflammatory in some chronic inflammatory conditions.8 It was previously shown that mROS release can stabilize HIF-1α in other cell types; however, it was unclear how this process is operational in neutrophils. Given that mitochondrial electron transport complexes have limited expression in neutrophils, it has been proposed that other pathways of mROS generation may be functional in these cells. Previously, an operational role for the G-3-P shuttle was described in neutrophils,9 similar to lymphocytes, where this shuttle links glycolysis and mitochondrial ROS synthesis.
Building on this previous work, Willson et al tested whether neutrophils would be able to stabilize HIF-1α through mROS release by using the G-3-P shuttle, and whether this pathway would link neutrophil glycolysis and HIF stabilization under conditions of hypoxia. They found that exposure of neutrophils to hypoxic in vitro conditions led to enhanced mROS synthesis, but not other sources of ROS. This phenomenon was associated with enhanced neutrophil survival through decreased apoptosis. Using inhibitors of mROS synthesis and of the electron transport chain, the authors found that these pathways were instrumental in stabilizing HIF-1α expression in response to hypoxia. Furthermore, G-3P dehydrogenase 2 (GPD2), the mitochondrial component of the G-3-P shuttle, was shown to be the molecule by which neutrophils produce mROS, stabilize HIF-1α, and promote neutrophil survival and other key effector functions under conditions of hypoxia. Furthermore, the authors found that modulation of glycolysis leading to increased flux through the G-3-P shuttle promotes enhanced mitochondrial membrane potential and mROS release during hypoxia.
Overall, the results from this study highlight a pathway promoting HIF-1 stabilization in neutrophils and improved cell survival during hypoxic conditions (see figure). This has implications for many diseases where low oxygen levels are prevalent, including pulmonary complications of SARS-CoV-2, where neutrophils are prominent pathogenic players in the lung tissue.10 Given the link between mROS and neutrophil extracellular traps (NETs)4 it will be interesting to evaluate how enhancement in mROS during hypoxia may modulate neutrophil functions beyond effects on apoptosis. The study has some limitations, including the use of cell lines (given how difficult it is to genetically manipulate primary neutrophils) and the lack of in vivo data that can support both the in vitro findings and the potential pharmacologic approaches that could be considered to modulate immunometabolism. Nevertheless, the results of the study expand our knowledge on the modulation of metabolic pathways in myeloid cells in conditions of low oxygen and have implications not only for acute infections, but also in chronic inflammatory conditions, such as rheumatoid arthritis and inflammatory bowel disease, where dysregulation of inflammatory and hypoxia response pathways may play important pathogenic roles.
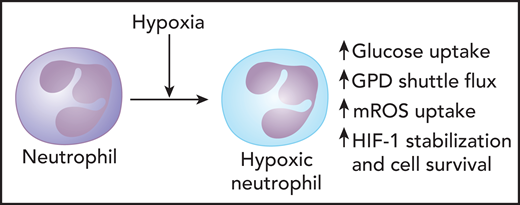
Mitochondria play important role in modulating metabolic responses of neutrophils during hypoxia. During hypoxic conditions, neutrophils synthesize enhanced mROS formation, which stabilizes HIF-1α, a transcriptional regulator that promotes oxygen delivery to hypoxic regions. This process relies on the G-3-P shuttle pathway. Modulation of glycolysis leading to increased flux through the G-3-P shuttle promotes enhanced mitochondrial membrane potential and mROS release during hypoxia. GPD2, the mitochondrial component of the G-3-P shuttle, is involved in promoting mROS synthesis, HIF-1α stabilization, and neutrophil survival during hypoxia. Professional illustration by Patrick Lane, ScEYEnce Studios.
Mitochondria play important role in modulating metabolic responses of neutrophils during hypoxia. During hypoxic conditions, neutrophils synthesize enhanced mROS formation, which stabilizes HIF-1α, a transcriptional regulator that promotes oxygen delivery to hypoxic regions. This process relies on the G-3-P shuttle pathway. Modulation of glycolysis leading to increased flux through the G-3-P shuttle promotes enhanced mitochondrial membrane potential and mROS release during hypoxia. GPD2, the mitochondrial component of the G-3-P shuttle, is involved in promoting mROS synthesis, HIF-1α stabilization, and neutrophil survival during hypoxia. Professional illustration by Patrick Lane, ScEYEnce Studios.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal