

In this issue of Blood, Das et al demonstrate that the intestinal deletion of NCOA4, a mediator of ferritin degradation, attenuates systemic iron loading in a mouse model of hemochromatosis.1
The absorption of dietary iron across the proximal small intestine is normally tightly controlled to prevent the deleterious effects of systemic iron deficiency and systemic iron overload. Nonheme iron enters enterocytes via the apical brush border transporter DMT1 and exits into the portal circulation via the basolateral membrane transporter ferroportin. The peptide hormone hepcidin, which hepatocytes secrete in response to increased body iron stores and other stimuli, regulates this process by limiting ferroportin-mediated iron efflux from enterocytes. Additionally, during states of iron deficiency, the hypoxia inducible factor-2α (HIF-2α) transcription factor promotes iron uptake by increasing expression of the DMT1 and ferroportin transporters.
How iron traffics through the enterocyte cytosol to reach the basolateral membrane is less well understood. Suggesting that the multimeric iron-storage protein ferritin participates in this process, mice with enterocyte-specific deletion of the ferritin heavy chain show reduced iron concentration in their intestines and develop systemic iron loading, even though their hepatic hepcidin expression is increased.2 In the present study, Das and colleagues increase our understanding of iron trafficking within enterocytes by clarifying the role of nuclear receptor coactivator 4 (NCOA4) in this cell type. NCOA4 is a ubiquitously expressed coiled-coil domain protein that promotes iron release from ferritin stores by facilitating the delivery of ferritin to lysosomes for degradation (a process termed “ferritinophagy”).3 Accordingly, mice with global Ncoa4 loss accumulate iron in multiple organs, including duodenum.4 Although tissue-specific NCOA4 depletion has revealed local roles for NCOA4 in iron handling by erythroid cells5 and hepatocytes,6 its function in enterocytes remained to be clarified.
Das and colleagues observed induction of NCOA4 messenger RNA (mRNA) and protein in mouse duodenum in response to diet-induced iron deficiency and chemically induced hemolytic anemia, 2 physiological perturbations that are known to activate local HIF-2α–mediated transcriptional responses in the duodenum. To directly implicate HIF-2α in NCOA4 regulation in enterocytes, the investigators used a series of complementary experiments in cultured cells and genetically engineered mouse models. Heterodimeric HIF transcription factors activate target gene expression by binding to specific hypoxia response elements (HREs) in DNA. Importantly, HIF levels increase under conditions of iron deficiency, as well as hypoxia, because the HIF prolyl hydroxylase (PHD) enzymes that mark HIF α subunits for degradation are iron- and oxygen-dependent enzymes. Das et al found that hypoxic exposure and iron chelation each raised NCOA4 mRNA in human intestinal organoids, and pharmacologic HIF activation, mediated by a PHD inhibitor, raised duodenal Ncoa4 mRNA in mice. However, mice lacking HIF-2α in the intestinal epithelium failed to upregulate duodenal NCOA4 in response to dietary iron deficiency. Mice with genetically induced HIF stabilization in enterocytes showed elevated levels of NCOA4 and ferritin heavy chain in duodenum, which were dependent upon the presence of HIF-2α. The authors also found that the Ncoa4 gene promoter, which contains an HRE, is a direct HIF-2α target in intestinal cells (see figure).
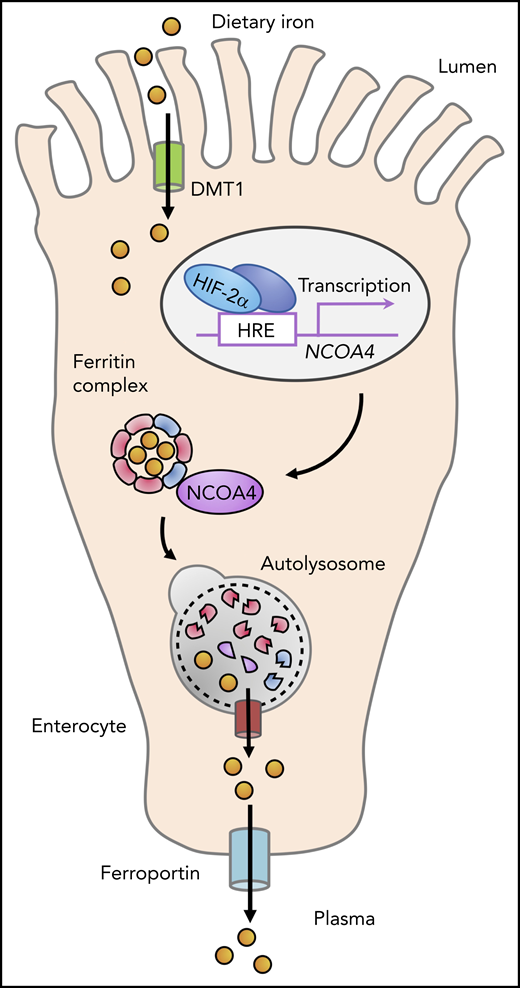
During the absorption of dietary nonheme iron in the proximal small intestine, iron traverses the enterocyte cytosol, where it may be stored in ferritin complexes. NCOA4, a cargo receptor that promotes the autophagic degradation of ferritin, is expressed in enterocytes and is upregulated by HIF-2α. NCOA4 activity enhances iron uptake in the context of systemic iron overload, because loss of NCOA4 in enterocytes attenuates systemic iron loading in a mouse model of hemochromatosis caused by hepcidin gene disruption.
During the absorption of dietary nonheme iron in the proximal small intestine, iron traverses the enterocyte cytosol, where it may be stored in ferritin complexes. NCOA4, a cargo receptor that promotes the autophagic degradation of ferritin, is expressed in enterocytes and is upregulated by HIF-2α. NCOA4 activity enhances iron uptake in the context of systemic iron overload, because loss of NCOA4 in enterocytes attenuates systemic iron loading in a mouse model of hemochromatosis caused by hepcidin gene disruption.
Next, to determine whether intestinal NCOA4 activity modulates iron absorption in the steady-state or under conditions of perturbed systemic iron balance, the investigators generated mice in which the Ncoa4 gene was disrupted specifically in enterocytes. Although these mice showed increased ferritin heavy chain expression in duodenum, compatible with impaired ferritinophagy in enterocytes, they retained normal hemoglobin, liver iron, and serum iron levels under basal conditions, and they showed hematological responses similar to controls when subjected to chemically induced hemolytic anemia. Interestingly, however, in a mouse model of iron overload caused by genetic ablation of hepcidin in hepatocytes, the investigators observed Ncoa4 mRNA upregulation in duodenum. Of note, a prior study of this hepcidin-deficient hemochromatosis model detected HIF-2α stabilization in the duodenum that was attributed to an intracellular iron-deficient state in enterocytes caused by increased ferroportin mediated iron efflux.7 Finally, the investigators used genetic approaches in mice to directly test whether the systemic iron loading caused by hepcidin gene disruption could be modulated by loss of NCOA4 in enterocytes. In response to hepcidin deficiency, mice that retained NCOA4 in enterocytes developed hyperferremia and iron accumulation in liver and pancreas, whereas mice lacking NCOA4 in enterocytes showed an attenuation of systemic iron loading that was accompanied by iron retention in the duodenum.
Collectively, these findings in mouse models suggest that NCOA4 activity in enterocytes enhances iron absorption to promote systemic iron loading in the hyperabsorption state caused by hepcidin deficiency (see figure). The fact that duodenal NCOA4 loss did not alter systemic iron parameters in mice with intact hepcidin regulation under basal conditions may point to a relatively low rate of ferritinophagy in the iron-balanced state. Indeed, although NCOA4 expression is transcriptionally upregulated by HIF in intestinal1 and hepatic cells,6 NCOA4 is also known to be posttranslationally downregulated under iron-replete conditions by the HERC2 E3 ubiquitin-protein ligase, which promotes NCOA4 degradation by the proteasome.8 Although Das et al did not detect a role for enterocyte NCOA4 in the setting of acute hemolytic anemia, additional studies of mice fed chronically with a low-iron diet, which the investigators found could induce duodenal NCOA4 expression, are needed to clarify whether NCOA4 activity in enterocytes modulates iron absorption in iron-deficient states.
In addition to clarifying the physiology of duodenal iron absorption, the present results suggest a novel therapeutic approach to limit iron uptake in clinical disorders characterized by the pathological hyperabsorption of iron. Hepcidin levels are inappropriately low relative to body iron stores in hereditary forms of hemochromatosis and in congenital iron-loading anemias, and a number of potential therapeutic approaches to limit systemic iron loading in these disorders are under investigation, including hepcidin agonists and ferroportin antagonists.9 The study of Das et al suggests local inhibition of NCOA4 activity in enterocytes as an alternative approach to limit iron uptake by trapping iron within ferritin in these cells. Such inhibition might be achieved by formulations that permit direct gastrointestinal tract delivery10 of small interfering RNAs targeting NCOA4 and potentially could be coupled with other investigational agents.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal