

In this issue of Blood, Korona and colleagues1 describe a novel mechanism involving GPR34 mutations linking the lymphoepithelial lesions of Sjögren syndrome to the development of MALT lymphoma.
In 1971 Azzopardi and Evans described the association of myoepithelial sialadenitis, also called benign lymphoepithelial lesion (LEL) of the parotid gland, with the subsequent development of malignant lymphoma.2 Today, the World Health Organization classification of lymphomas recognizes 3 types of marginal zone lymphomas (MZLs): nodal, splenic, and extranodal MZLs of the mucosa associated-lymphoid tissues (MALT). MALT lymphomas are indolent B-cell non-Hodgkin lymphomas that often arise on a background of chronic microbial infection or autoimmune disorders at diverse extranodal sites in organs that physiologically are devoid of lymphoid tissue.3 The most frequent and best characterized is MALT lymphoma of the stomach, associated with chronic Helicobacter pylori gastritis.4 In contrast, MALT lymphomas affecting the salivary glands and the thyroid develop in the context of chronic inflammation and lymphoid hyperplasia secondary to autoimmune disorders, such as Sjögren syndrome and Hashimoto thyroiditis, respectively. Regardless of the cause of chronic inflammation, it is now recognized that this constant antigenic stimulation induces B-cell receptor (BCR) signaling and CD40 receptor activation in B cells, leading to NF-κB pathway activation, which triggers local proliferation of B cells that eventually undergo malignant transformation through gaining genetic alterations.5 MZLs have in common recurrent genetic aberrations that constitutively activate the NF-κB pathway. The most frequent oncogenic event is the inactivation by mutations or deletions of the TNFAIP3 (A20) gene, a negative regulator of NF-κB signaling. The characteristic translocations t(11;18)(q21;q21), t(14;18)(q32;q21), and t(1;14)(p22;q32) that deregulate MALT1, BCL10, and BIRC3 are exclusively found in MALT lymphomas and trigger NF-κB activation independent of antigen stimulation. In addition, there is a significant site dependence of the mutational profiles and translocation frequencies of MALT lymphomas, probably reflecting the impact of different etiologies and local microenvironments. Accordingly, NOTCH2, PTPRD, TBL1XR1, and KLF2 mutations are more specific to the nodal and splenic MZLs, but similarly result in chronic BCR stimulation and further induce downstream NF-κB activation. More recently, recurrent somatic mutations in the G-protein–coupled receptor 34 (GPR34), not previously described in human neoplasias, were identified in 16% of MALT lymphomas of the salivary gland providing further evidence for the cooperation between immune receptor signaling and mutations in MALT lymphoma pathogenesis.6
GPR34 is a member of class A G-protein–coupled receptor (GPCR) superfamily that, through G-protein activation, transduces extracellular stimuli into intracellular signals. G-protein activation promotes the intracellular changes that lead to biological responses.7 Upon ligand binding, GPCRs undergo conformational changes to bind and activate heterotrimeric G-proteins at the cell membrane that in turn regulate downstream signaling targets. These receptors are closely regulated by phosphorylation in a motif localized in the C-terminal intracytoplasmic tail in most GPCRs that are responsible for interactions with β-arrestin. The binding of GPCR to β-arrestin triggers the receptor internalization and dampening of intracellular signaling, a process designated desensitization.7 Interestingly, most GPR34 mutations in salivary gland MALT lymphomas are nonsense or frameshift indels predicted to impair or delete the phosphorylation sites in the C-terminal region and therefore deregulate desensitization of the receptor.6
In the current study Korona et al, using GPR34 expression constructs, elegantly demonstrated that GPR34 mutations observed in salivary gland MALT lymphomas, especially the Q340X truncation in the C-terminal region, confer increased resistance to apoptosis and substantially increased transformation potential. The resulting mutant also has a significantly delayed internalization after ligand stimulation and therefore remains constitutively activated and is capable of activating diverse signaling pathways critical for cellular function, such as NF-κB and AP1 (MAPK/JNK). Most importantly, Korona et al discovered a potential mechanism of paracrine stimulation of malignant B-cells via GPR34 through its ligand lysophosphatidylserine generated by the unique microenvironment of LELs, independent of GPR34 mutation status. Indeed, mutated or overexpressed GPR34 is more sensitive to ligand stimulation and enhances its activation. An interesting aspect of the study was to demonstrate that phospholipase A1 (PLA1), expressed by ductal epithelium of normal salivary glands as part of its exocrine secretion, catalyzes the generation of lysophosphatidylserine from phosphatidylserine exposed on apoptotic cells present in LELs. Furthermore, the presence of local PLA1 may explain the organ tropism of GPR34 activation and mutations. The model suggests that LELs forming in response to chronic inflammation undergoes continuous epithelial cell regeneration and apoptosis with increased ligand production perpetuating the stimulation of GPR34 in B-cells that eventually will undergo malignant transformation and acquire genetic aberrations (see figure). This important observation demonstrates that organ- and disease-specific chronic inflammatory stimuli and acquired genetic alterations play key roles in MALT lymphoma pathogenesis by dysregulating similar molecular mechanisms. Furthermore, this work is a nice example of how the investigation of the functional consequences of a mutation can lead to insights into more general pathogenetic mechanisms, in this case the generation of a paracrine stimulus for tumor growth through interaction with a specific inflammatory microenvironment.
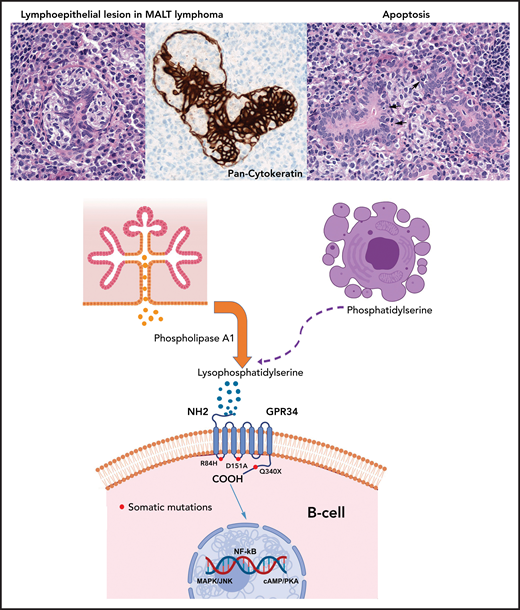
Paracrine GPR34 activation in salivary gland MALT lymphoma. Salivary gland MALT lymphoma (top) with typical lymphoepithelial lesion (left) highlighted by pan-cytokeratin staining (center) and increased apoptotic bodies (right, arrows). Phospholipase A1 is normally secreted by salivary gland epithelia (center left) and catalyzes generation of lysophosphatidylserine from phosphatidylserine exposed on apoptotic cells (center right). Lysophosphatidylserine binds to GPR34 and activates downstream signaling through the NF-κB, MAPK/JNK and cAMP/PKA pathways. The paracrine stimulation is independent of GPR34 mutational status. Red dots mark the location of the GPR34 mutations investigated by Korona et al, with the Q340X mutation in the C-terminal tail showing the most pronounced effects followed by the D151A mutation. The R84H mutation showed an effect similar to that of the GPR34 wild-type.
Paracrine GPR34 activation in salivary gland MALT lymphoma. Salivary gland MALT lymphoma (top) with typical lymphoepithelial lesion (left) highlighted by pan-cytokeratin staining (center) and increased apoptotic bodies (right, arrows). Phospholipase A1 is normally secreted by salivary gland epithelia (center left) and catalyzes generation of lysophosphatidylserine from phosphatidylserine exposed on apoptotic cells (center right). Lysophosphatidylserine binds to GPR34 and activates downstream signaling through the NF-κB, MAPK/JNK and cAMP/PKA pathways. The paracrine stimulation is independent of GPR34 mutational status. Red dots mark the location of the GPR34 mutations investigated by Korona et al, with the Q340X mutation in the C-terminal tail showing the most pronounced effects followed by the D151A mutation. The R84H mutation showed an effect similar to that of the GPR34 wild-type.
Besides GPR34 mutations, deregulation of GPR34 has been reported in salivary gland MALT lymphomas as a consequence of a rare recurrent chromosomal translocation t(X;14)(p11;q32) that juxtaposes the GPR34 gene to the enhancers of the immunoglobulin heavy chain gene.8,9 Nevertheless, elevated GPR34 mRNA expression was detected in most MALT lymphomas analyzed, regardless of the presence of the translocation indicating a functional role of this GPCR receptor in lymphomagenesis.9 The role of GPR34 activation in MALT lymphomas in other organs, as well as in B-cell lymphomas in general, should be investigated to see whether receptor activation through similar mechanisms plays a role in their pathogenesis. Importantly, GPCR pharmacological targeting has shown the potential of modulating these receptors for the development of novel anticancer therapeutics10 that open new treatment avenues for patients with Sjögren syndrome and salivary gland MALT lymphomas.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal