

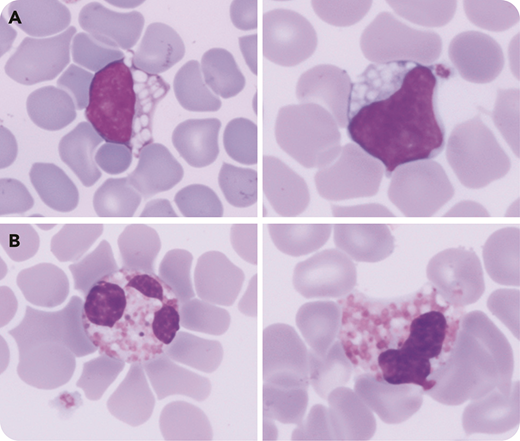
A routine blood count was performed in a premature boy (35 weeks’gestation) at day 2 of life revealing only a mild thrombocytopenia (platelets, 145 × 109/L) associated with an abnormal lymphocytes/blasts alarm (Sysmex XN-3000). Examination of a May-Grünwald Giemsa–stained blood smear showed numerous lymphocytes with large vacuoles (75%) (panel A, original magnification ×1000) associated with hypogranulated eosinophils and variable tinctorial affinity (panel B, original magnification ×1000). In general, such atypical lymphocytes are evocative of lysosomal storage disorder, but their association with these abnormal eosinophils is highly specific of GM1 gangliosidosis (Landing disease). Despite the absence of clinical signs or family history (consanguinity), this cytology encouraged clinicians to investigate further. A urine sample revealed GM1 ganglioside elevation with traces of GM2 ganglioside. Biochemical investigation and molecular analysis confirmed type 1 GM1 gangliosidosis with total enzymatic deficiency of β-galactosidase in leukocytes, as well as an undescribed homozygous mutation (c.819delG) on GLB1.
GM1 gangliosidosis is a rare fatal disease with no curative treatment, and is usually diagnosed at 8 months with the onset of symptoms that are caused by ganglioside deposits in various tissues. Prebirth or perinatal diagnoses have been reported, but are usually accompanied by severe symptoms or family history. To our knowledge, we present the first case of type 1 GM1 gangliosidosis diagnosed at day 2 of life that was strongly suspected based on a careful cytological analysis without clinical symptoms or family history.
A routine blood count was performed in a premature boy (35 weeks’gestation) at day 2 of life revealing only a mild thrombocytopenia (platelets, 145 × 109/L) associated with an abnormal lymphocytes/blasts alarm (Sysmex XN-3000). Examination of a May-Grünwald Giemsa–stained blood smear showed numerous lymphocytes with large vacuoles (75%) (panel A, original magnification ×1000) associated with hypogranulated eosinophils and variable tinctorial affinity (panel B, original magnification ×1000). In general, such atypical lymphocytes are evocative of lysosomal storage disorder, but their association with these abnormal eosinophils is highly specific of GM1 gangliosidosis (Landing disease). Despite the absence of clinical signs or family history (consanguinity), this cytology encouraged clinicians to investigate further. A urine sample revealed GM1 ganglioside elevation with traces of GM2 ganglioside. Biochemical investigation and molecular analysis confirmed type 1 GM1 gangliosidosis with total enzymatic deficiency of β-galactosidase in leukocytes, as well as an undescribed homozygous mutation (c.819delG) on GLB1.
GM1 gangliosidosis is a rare fatal disease with no curative treatment, and is usually diagnosed at 8 months with the onset of symptoms that are caused by ganglioside deposits in various tissues. Prebirth or perinatal diagnoses have been reported, but are usually accompanied by severe symptoms or family history. To our knowledge, we present the first case of type 1 GM1 gangliosidosis diagnosed at day 2 of life that was strongly suspected based on a careful cytological analysis without clinical symptoms or family history.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal