

In this issue of Blood, report the efficacy of hematopoietic stem cell transplantation (HSCT) for children with a rare immune deficiency and autoinflammation disorder termed POMP-related autoinflammation and immune dysregulation (PRAID).1
A large majority of HSCTs worldwide are performed in adults with bone marrow malignancy.2 The decision to perform such transplantations is typically informed by a significant body of experience indicating appropriate use of transplantation and, in the best of circumstances, data from well-designed and -conducted clinical trials. The smaller numbers of children undergoing HSCT and the diversity of indications mean that data are often much more limited in this setting, and determining appropriateness of HSCT can be difficult. Children with immune deficiency and dysregulation disorders present a particular challenge. In many disorders, there is significant phenotypic diversity, and the severity of the phenotype may be poorly predicted by laboratory testing. Also, reasonably effective supportive care is available for many disorders. Lastly, and perhaps most troublingly, in very infrequent disorders, it may be unclear which, if any, parts of the phenotype may be ameliorated by HSCT. Taken together, these variables can make the decision to proceed to transplantation in a small child with an incompletely understood immune disorder agonizingly difficult for both parents and physician, and the consequences of a regretted decision can be severe.
Immune dysregulatory disorders are particularly complex in determining the likelihood of improvement with HSCT, because restoration of completely normal immunity after HSCT may require interaction with host tissues that will not be replaced by allogeneic hematopoietic stem cells.3 PRAID is an example of a group of disorders known as proteosome associated autoinflammatory syndromes, and POMP is a chaperone for proteasome assembly.4 Proteasomes perform a range of key cellular functions, including the regulation of protein homeostasis, major histocompatibility class I antigen processing, cell-cycle proliferation, and signaling. Proteasomal defects generally lead to a markedly increased type 1 interferon signature, clinically manifested as autoinflammation, which can be ameliorated by immune suppression, and infections resulting from ineffective immunity, which are worsened by immune suppression, presenting a significant therapeutic struggle.
Martinez et al performed transplantation in 2 children with PRAID, both of whom faced a difficult-to-resolve conflict between the need to provide immune suppression for autoinflammation and the need to avoid immune suppression because of life-threatening infections (see figure). They report normalization of CD8+, CD4+, and memory T-cell proportions and restoration of T-cell cytokine secretion, with improved diversity of the T-cell repertoire after HSCT. Most importantly, the children experienced significant clinical benefit, with resolution of both autoinflammation and severe infections, and both are surviving, almost 3 and 5 years after HSCT, respectively.
This report is an example of 1 case or a small number of cases importantly informing both clinical practice and our understanding of the biology of a disease. A physician might have reasonably been concerned that T-cell recovery would have been limited by persistent proteasome deficiency in the cortical thymic epithelium, but this was not observed, which might reflect repopulation of the thymus with donor dendritic cells after HSCT. Overexpression of type 1 interferon–regulated genes is key to the abnormal phenotype, and encouragingly, downregulation of the dysregulated interferon response was seen after HSCT, indicating that most or all of the systemic interferonopathy originated from immune cells.
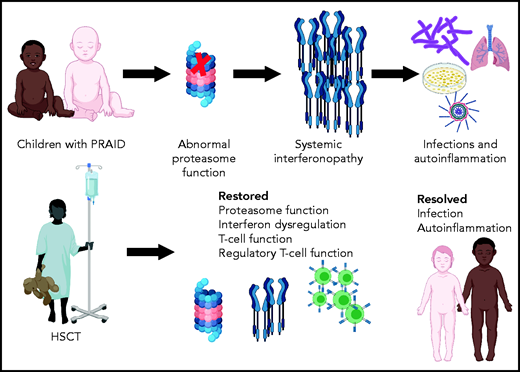
Taken together, these findings indicate that HSCT can be an appropriate and effective treatment of at least 1 proteasome-associated autoinflammatory disorder. HSCT is a new therapeutic strategy that might advance management of these disorders, to be added to recent new additions of Jak1/Jak2 inhibition with ruxolitinib and infusion of emapalumab to inhibit interferon production.5,6 The often partial efficacy of these new nontransplantation strategies, and others that might be developed, will now need to be weighed against the risk of severe organ damage from autoinflammation or of a life-threatening infection intervening before HSCT can be performed. Despite the survival of both children reported by Martinez et al, there is an expected mortality of at least 10% for children with severe immune disorders undergoing HSCT.7 Moreover, both children in this study developed both acute and chronic graft-versus-host disease (GVHD), which was fortunately highly responsive to steroids, but with any allogeneic HSCT performed for a chronic nonmalignant condition, there is the possibility of exchanging 1 chronic disorder for another (ie, chronic GVHD). As the authors point out, the risk of GVHD might be reduced by interferon blockade during the acute phase of transplantation. Although the availability of a new therapeutic modality for children with proteasome disorders is highly welcome, the decision of whether to use a therapy with significant risk of immediate mortality will remain difficult for all concerned.
Conflict-of-interest disclosure: S.M.D. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal