

In this issue of Blood, have further developed gene editing approaches to increasing fetal hemoglobin (HbF) expression in vitro and in a murine model with engrafted human CD34+ thalassemic cells.1 This may improve outcomes in the future for patients with severe inheritable hemoglobin disorders.
Inherited red cell function disorders represent a major global health care burden as a result of the chronic multisystem nature of these diseases and the associated long-term side effects of conventional treatment strategies for those with severe forms of the disease.2
In the majority of individuals at birth, there is a switch from the production of HbF (α2γ2) to adult hemoglobin (α2β2). However, in a minority, this switch is incomplete and results in the persisting expression of HbF (hereditary persistence of HbF). This is a benign condition, and it has long been recognized that the continuing coexisting expression of HbF in patients with β hemoglobinopathies, such as sickle cell disease or β-thalassemia, can reduce the severity of the disease. There have been a number of breakthroughs in our understanding of the mechanisms of gene regulation at the HBG locus in recent years (see figure). These advances include the identification of transcription factors BCL11A and LRF (also known as ZBTB7A), which play key roles in suppressing transcription of γ-globin by direct binding at the HBG promoter.3-5 LRF/ZBTB7A and BCL11A appear to bind to 2 specific sequences that are 200 bp and 115 bp upstream of the HBG promoter (hereafter referred to as HBG-200 and HBG-115). Gene editing technology to modulate the expression of HbF, through the disruption of these transcriptional regulatory pathways in hematopoietic stem and progenitor cells (HSPCs),3-6 is now being evaluated in early-phase clinical trials. These efforts have largely focused on mobilizing HSPCs, followed by ex vivo clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 editing. The most common strategy involved downregulating BCL11A expression using CRISPR-Cas9 editing with a single guide RNA (sgRNA) that targets tissue-specific enhancers that regulate the expression of BCL11A in erythroid cells.7
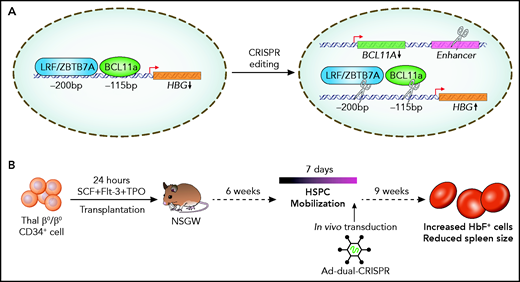
Regulation of γ globin production in humans. (A) At birth, LRF/ZBTB7A and BCL11A bind at cis regulatory regions at HBG (−200 bp and −115 bp from the promoter) to repress γ globin production. Combined CRISPR-Cas9 editing of the BCL11A enhancer, and LRF/ZBTB7A or BCL11A binding sites, to derepress γ globin production. (B) In vivo transduction of human CD34+ cells, from patients with β-thalassemia engrafted in NSGW mice, with a dual sgRNA–containing adenoviral vector.
Regulation of γ globin production in humans. (A) At birth, LRF/ZBTB7A and BCL11A bind at cis regulatory regions at HBG (−200 bp and −115 bp from the promoter) to repress γ globin production. Combined CRISPR-Cas9 editing of the BCL11A enhancer, and LRF/ZBTB7A or BCL11A binding sites, to derepress γ globin production. (B) In vivo transduction of human CD34+ cells, from patients with β-thalassemia engrafted in NSGW mice, with a dual sgRNA–containing adenoviral vector.
In this study, the investigators aimed to increase the applicability and efficacy of this strategy by targeting different combinations of these regulatory elements using CRISPR-Cas9 editing and by simplifying the delivery of sgRNA and Cas9 enzyme through the use of an adenoviral vector in vivo. First, they compared the relative efficiency of editing the HBG-200 locus vs editing the BCL11A enhancer or the HBG-115 locus in reactivating HbF. Having identified sgRNAs for the HBG-200 locus that efficiently increased HbF expression, they used multiplex sgRNA that targeted different combinations of the BCL11A enhancer or the HBG-200 or HBG-115 loci in CD34+ cells to systematically compare the relative merits of each approach. This demonstrated that combining BCL11A enhancer editing with either of the HBG loci resulted in an increased frequency of HbF+ cells and total γ globin levels.
Because the ultimate goal of such technology is to establish long-lived progenitors, the investigators could show that this gene editing approach did not lead to a reduction in the clonogenic potential of edited HSPCs. It should also be noted that they did not see any clear evidence of an increase in off-target DNA damage, which is a recurring concern in this field. Furthermore, human HSPCs doubly edited at the BCL11A enhancer and HBG-115 were able to engraft in vivo and stably reconstitute human hematopoiesis in a recipient mouse.
Current gene therapy approaches require harvesting cells from patients, followed by ex vivo genome editing. The genetically modified cells are then transplanted back into patients using conditioning chemotherapy. Therefore, an in vivo CRISPR-Cas9 editing strategy has significant benefits. In this study, the team further optimized a nonintegrating adenoviral vector that can now contain 2 sgRNA. This was able to successfully edit primary CD34+ HSPCS from thalassemic patients and resulted in an improvement in erythroid differentiation. In a murine model engrafted with human CD34+ thalassemic cells, they were able to successfully reactivate HbF production by mobilizing stem cells into the peripheral circulation, followed by in vivo transduction with the dual sgRNA–containing adenoviral vector (see figure). Engraftment of human CD45+ thalassemic cells in secondary transplantation assays suggests that editing of true hematopoietic stem cells was achieved.
Of course, a number of obstacles remain before this work can be translated into the clinic and impact patients. The investigators note that a particular caveat to their strategy is the immunogenicity of using adenoviral vectors, although this is likely to be ameliorated by rapidly developing viral vector technology. Nonetheless, the investigators should be congratulated on developing a potentially important strategy in improving HbF production in patients with severe defective hemoglobin phenotypes.
Conflict-of-interest disclosure: J.L. has received travel funding from Novartis and Daichi-Sankyo and has received honoraria from Pfizer, Janssen Pharmaceuticals, and Amgen.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal