

In this issue of Blood, 1 identified a direct causative link between interleukin-18 (IL-18), arrhythmic burden, and myocardial fibrosis in the cardiomyopathy seen with sickle cell disease (SCD). In SCD, cell sickling leads to ischemia and inflammation in the myocardium, producing diffuse myocardial fibrosis2 and resulting in diastolic dysfunction and cardiomyopathy.3 Treatment of SCD with blood transfusions may exacerbate cardiac dysfunction because of an increase in inflammation with a smaller risk of iron-mediated injury owing to elevation of nontransferrin bound iron and labile plasma iron. In animal models, iron-mediated injury results in diastolic and systolic dysfunction with increased oxidative stress and myocardial fibrosis.4 Thus, heart disease associated with SCD is driven by chronic anemia, microvascular disease, systemic inflammation, and iron-mediated injury.
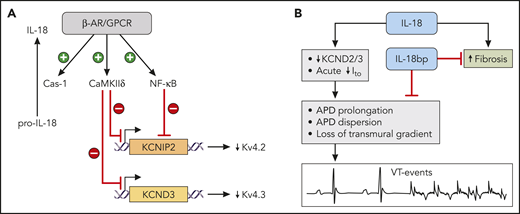
IL-18 in heart failure and SCD. (A) Control of IL-18 and Kv 4.x channels by β-adrenergic signaling (β-AR/GPCR) via CaMKIIδ and NF-κB pathways. These pathways inhibit expression of KCNIP2 (the gene for chaperon protein for Kv 4.2) and KCND3 (the gene for Kv 4.3). (B) Role of IL-18 in promoting arrhythmias and fibrosis in SCD cardiomyopathy. The adverse effects of IL-18 can be counteracted in the murine model by the treatment with IL-18bp for 4 weeks. APD, action potential duration. Professional illustration by ScEYEnce Studios.
IL-18 in heart failure and SCD. (A) Control of IL-18 and Kv 4.x channels by β-adrenergic signaling (β-AR/GPCR) via CaMKIIδ and NF-κB pathways. These pathways inhibit expression of KCNIP2 (the gene for chaperon protein for Kv 4.2) and KCND3 (the gene for Kv 4.3). (B) Role of IL-18 in promoting arrhythmias and fibrosis in SCD cardiomyopathy. The adverse effects of IL-18 can be counteracted in the murine model by the treatment with IL-18bp for 4 weeks. APD, action potential duration. Professional illustration by ScEYEnce Studios.
The inflammatory biomarker, IL-18, is an established biomarker of heart disease. In patients, elevated plasma levels of IL-18 are associated with disease progression for coronary artery disease and with increased mortality for advanced heart failure.5 In patients with atrial fibrillation (AF) without structural heart disease, IL-18 is independently associated with AF, and IL-18 levels are higher in patients with persistent AF than in patients with paroxysmal AF.6 Current understanding of the link between arrhythmias and elevated levels of IL-18 is that both are a reflection of upregulated activity of the β-adrenergic signaling pathway (β-adrenergic receptor and other related G protein–coupled receptors, β-AR/GPCR) in heart disease (see figure panel A). Activation of β-adrenergic signaling pathway promotes cleavage of pro-IL-18 by caspase-1 via inflammasome activation (see figure, panel A).7 At the same time, the pathway also activates calmodulin (CaMKIIδ) and nuclear factor κB (NF-κB) pathways, which in turn suppress the expression of KCNIP2 (the gene for the chaperone protein of Kv 4.2) and KCND3 (the gene for Kv 4.3) (see figure panel A), resulting in lower density of transient outward current (Ito).8 This reduction in repolarizing Ito produces a higher risk of arrhythmia via 2 mechanisms. First, reduction in repolarizing Ito leads to prolongation of action potential duration (APD). Second, the reduction of Ito diminishes a transmural gradient of Ito (higher density in the epicardium than in the endocardium), which prevents transmural reentry.9 This framework provided a reasonable explanation of the link between IL-18 plasma levels and elevated risk of arrhythmias in patients and animal models. Gupta et al expand this framework by providing the direct causative link between IL-18, arrhythmic burden, and myocardial fibrosis in an animal model SCD cardiomyopathy. In these settings, administration of IL-18 reduced Ito in the cardiomyocytes from SCD hearts, and exposure of isolated hearts to IL-18 prolonged APD and increased dispersion of APD, resulting in higher instances of arrhythmic events, such as premature ventricular contractions and ventricular tachycardia (VT) (see figure, panel B). The reverse intervention (treatment of mice with IL-18 binding protein, IL-18bp, for 4 weeks) was able to markedly reduce plasma IL-18 levels and NF-κB phosphorylation. These molecular changes were associated with reduced myocardial fibrosis, diminished VT burden, and reversal of left ventricular dilation and diastolic dysfunction (see figure panel B). These findings confirm the specificity of IL-18 involvement in arrhythmias and cardiac remodeling in the model of SCD. These results were correlated with clinical findings. SCD patients with lower IL-18 plasma levels had shorter QTc intervals, and the SCD patients with lower IL18 expression in peripheral blood mononuclear cells had shorter QTc and better 4-year survival rate. However, Gupta et al did not address the effect of IL-18 on oxidative stress and microvascular dysfunction.
Dissecting the role of IL-18 in cardiovascular disease has a twofold importance. On the one hand, anti–IL-18 therapies have a potential in treating cardiovascular diseases, and a trial of the safety and tolerability of an IL-18 antibody has been conducted (#NCT01035645) with early promising results.10 These results suggest that blocking IL-18 may have therapeutic effects in patients with SCD at risk for cardiac arrhythmias and adverse outcomes. On the other hand, IL-18 has been proposed as a component of anticancer treatment (#NCT00659178), and such use of IL-18 creates a potential risk of cardiac fibrosis and arrhythmias, creating a need for possible mitigating strategies and adjuvant therapies. These results illustrate the important and complex relationship between the immune system and heart disease and the potential of targeting this relationship as novel therapies for heart disease.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal