

In this issue of Blood, 1 investigate the role of B-cell activating factor (BAFF) in skewing the B-cell pool in chronic graft-versus-host disease (cGVHD) in a major histocompatibility mismatch mouse model. They found a previously unknown mechanism by which BAFF elevations lead to B-cell dysregulation in cGVHD. The investigators first confirm that BAFF levels are increased in the model, as has been described in preclinical models and clinical graft-versus-host-disease (GVHD).2,3 This elevation is due in part to increased production by recipient fibroblastic reticular cells early after hematopoietic stem cell transplantation (HSCT), and, subsequently by donor hematopoietic cells, including alloreactive CD4+ T follicular helper (TFH)-like cells that are increased in the cGVHD setting.4 Using BAFF-knockout and BAFF-transgenic mice, the investigators then show that BAFF increases an extrafollicular GL7+ B-cell subset5 by inducing its NOTCH2 upregulation. The latter occurs as a result of high BAFF levels, in concert with in vivo alloantigen stimulation. This particular mouse B-cell subset is likely the functional counterpart of the CD27+ circulating B-cell subset that is elevated in patients with cGVHD and is capable of sustained immunoglobulin G production.3 Unlike other B-cell subsets, GL7+ B cells appear to persist and resist activation-induced death that leads to the profound general B-cell lymphopenia that is characteristic of cGVHD.
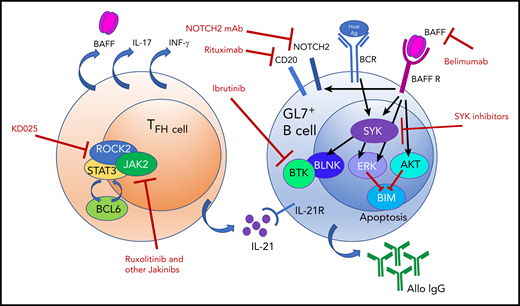
BAFF elevations and alloantigen stimulation promote a hyperresponsive GL7+ B-cell subset in cGVHD by stabilizing SYK after BCR signaling and inducing NOTCH2 expression. BAFF produced by follicular reticular cells (not shown) and TFH cells has multiple effects on GL7+ B cells, which result in hyperresponsiveness to BCR signaling. Targets for therapeutic intervention are shown and may perhaps be synergistically tested for clinical efficacy. Ag, antigen; AKT, AKT kinase; Allo IgG, allogeneic immunoglobulin G; BAFF R, BAFF receptor; BCL6, B-cell lymphoma 6 protein; BCR, B-cell receptor; BIM, Bcl-2 interacting mediator of cell death; BLNK, B-cell linker protein; BTK, Bruton’s tyrosine kinase; ERK, extracellular signal-regulated kinase; IFN-γ, interferon-γ; IL-17, interleukin-17; IL-21, interleukin-21; IL-21R, interleukin-21 receptor; Jakinibs, JAK inhibitors; KD025, Kadmon therapeutic agent 025 (orally available ROCK2 inhibitor); mAb, monoclonal antibody; ROCK2, Rho-associated kinase 2; SYK, spleen tyrosine kinase.
BAFF elevations and alloantigen stimulation promote a hyperresponsive GL7+ B-cell subset in cGVHD by stabilizing SYK after BCR signaling and inducing NOTCH2 expression. BAFF produced by follicular reticular cells (not shown) and TFH cells has multiple effects on GL7+ B cells, which result in hyperresponsiveness to BCR signaling. Targets for therapeutic intervention are shown and may perhaps be synergistically tested for clinical efficacy. Ag, antigen; AKT, AKT kinase; Allo IgG, allogeneic immunoglobulin G; BAFF R, BAFF receptor; BCL6, B-cell lymphoma 6 protein; BCR, B-cell receptor; BIM, Bcl-2 interacting mediator of cell death; BLNK, B-cell linker protein; BTK, Bruton’s tyrosine kinase; ERK, extracellular signal-regulated kinase; IFN-γ, interferon-γ; IL-17, interleukin-17; IL-21, interleukin-21; IL-21R, interleukin-21 receptor; Jakinibs, JAK inhibitors; KD025, Kadmon therapeutic agent 025 (orally available ROCK2 inhibitor); mAb, monoclonal antibody; ROCK2, Rho-associated kinase 2; SYK, spleen tyrosine kinase.
Furthermore, the investigators found that BAFF promotes sustained B-cell receptor (BCR)-driven activation of the aberrant B cells by increasing the half-life of the tyrosine kinase spleen tyrosine kinase (SYK). Normally, SYK is degraded upon BCR engagement to dampen BCR responsiveness. In the setting of cGVHD and elevated BAFF, pathogenic B cells maintain high SYK levels following BCR engagement (see figure). High levels of soluble BAFF, together with overall B-cell lymphopenia, promote the GL7+ subset, which does not undergo cell death upon encountering antigen. The persistence of this B-cell subset and likely its anti-host antibody production, not yet conclusively proven to be attributable to the GL7+ subset, correlate with severe cGVHD manifestations.
B-cell dysfunction has been implicated in autoimmune diseases and, in the setting of alloimmunity following HSCT,6 with cGVHD. Targeting of B cells has shown efficacy in the clinical treatment of GVHD,7-9 and ibrutinib is now approved as a second-line therapy for steroid-resistant cGVHD. Currently, Bruton’s tyrosine kinase (BTK) and SYK targeting is being tested in HSCT clinical trials. In their article, the investigators describe the role of BAFF in promoting a pathogenic BCR hyperresponsive subset in the preclinical cGVHD model. These exciting new data provide the field with further biological insight into the sustained activity of alloreactive B cells in cGVHD and indicate that therapeutic targeting of aberrant BAFF elevations could yield another potential treatment for cGVHD. Clinical testing of this approach is ongoing, and this body of work provides the rationale for BAFF targeting in GVHD and a model system to dissect the effects of the latter. Additionally, one could envision a combinatorial approach for GVHD using a BAFF inhibitor in conjunction with B- and T-cell–directed therapy, including BTK and SYK inhibitors. Additionally, because TFH cells are a major source of BAFF, targeting of this subset with a Rho-associated kinase 2 inhibitor,10 in combination with a BAFF inhibitor, may provide synergy. The refined understanding of how to reduce pathogenic B cells in cGVHD is another step forward in treating this prevalent and often morbid complication of HSCT.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal