

Key Points
RhoG deficiency abrogates cytotoxic function of CTLs and NK cells, causing HLH.
RhoG interacts with Munc13-4 at the surface of cytotoxic granules to promote their anchoring to the plasma membrane.

Abstract
Exocytosis of cytotoxic granules (CG) by lymphocytes is required for the elimination of infected and malignant cells. Impairments in this process underly a group of diseases with dramatic hyperferritinemic inflammation termed hemophagocytic lymphohistiocytosis (HLH). Although genetic and functional studies of HLH have identified proteins controlling distinct steps of CG exocytosis, the molecular mechanisms that spatiotemporally coordinate CG release remain partially elusive. We studied a patient exhibiting characteristic clinical features of HLH associated with markedly impaired cytotoxic T lymphocyte (CTL) and natural killer (NK) cell exocytosis functions, who beared biallelic deleterious mutations in the gene encoding the small GTPase RhoG. Experimental ablation of RHOG in a model cell line and primary CTLs from healthy individuals uncovered a hitherto unappreciated role of RhoG in retaining CGs in the vicinity of the plasma membrane (PM), a fundamental prerequisite for CG exocytotic release. We discovered that RhoG engages in a protein–protein interaction with Munc13-4, an exocytosis protein essential for CG fusion with the PM. We show that this interaction is critical for docking of Munc13-4+ CGs to the PM and subsequent membrane fusion and release of CG content. Thus, our study illuminates RhoG as a novel essential regulator of human lymphocyte cytotoxicity and provides the molecular pathomechanism behind the identified here and previously unreported genetically determined form of HLH.
Introduction
Cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells play key roles in host defense against intracellular pathogens and malignant cells. Complete or partial ablation of their cytotoxicity caused by impaired cytotoxic granule (CG) exocytosis underlies immunopathology of hemophagocytic lymphohistiocytosis (HLH) and HLH-associated syndromic diseases1,2 and may predispose to cancer.3-5 Mutations in the genes PRF1, UNC13D, STXBP2, and STX11 (encoding perforin, Munc13-4, Syntaxin-11, and Munc18-2, respectively6-10 ), selectively disrupting the pathway of perforin-mediated cytotoxicity in human lymphocytes, represent 4 genetic subtypes of classical, primary, or familial HLH (FHL2-5).11,12 At transient cytotoxic lymphocyte immune synapses (ISs), Syntaxin-11 is bound by Munc18-2 and actively recruited to the IS through the directed fusion of recycling endosomes with the plasma membrane (PM).13 Signals from activating receptors recruit Munc13-4 to CGs,14 where it interacts with Rab27a and promotes CG docking to the PM.15,16 A unique feature of Munc13-4 is that it lacks the archetypical membrane-targeting C1 domain of neuronal Munc13 isoforms.17 It is therefore unclear how Munc13-4 facilitates docking of CGs to the PM of cytotoxic lymphocytes. Furthermore, efficient CG release requires rearrangement and local clearance of cortical actin at the IS.18,19 However, how the activation of the exocytosis machinery and the remodeling of the actin cytoskeleton are coordinated in cytotoxic lymphocytes is poorly defined.
Here, we identified biallelic deleterious mutations in the gene encoding the small GTPase RhoG as a novel etiology of HLH. Our mechanistic characterization of this novel germline-encoded disease entity uncovers an essential role of RhoG in CG docking to the PM of cytotoxic lymphocytes.
Methods
Patient and ethics
The index patient was enrolled in the ongoing FINPIDD study series, with the study protocol reviewed and approved by the Ethics Committee of Helsinki University Hospital. Both parents gave written informed consent for the FINPIDD study. Patient studies have been approved by the Regional Ethical Review Board in Stockholm (study number 2013/1723-31/4) and the Ethical Committee of the Medical University of Vienna (EK499/2011).
Whole-exome sequencing
Whole-exome enrichment of the index case from a family of Finnish origin with HLH/FLH was performed using a SeqCap EZ MedExome Probes Kit (Roche Ltd, Basel, Switzerland). The sequencing was performed using HiSeq 1500 Rapid Run (Illumina, San Diego, CA) at the Institute for Molecular Medicine Finland Technology Centre, University of Helsinki, as previously described.20,21
Flow cytometric analyses
Immunophenotyping was performed using standard techniques. In brief, peripheral blood mononuclear cells from the patients, parents, and healthy donors were isolated using Ficoll density gradient centrifugation and stained cells were analyzed using BD LSR Fortessa, BC FACS Canto, or BD FACS Calibur. Fluorescence-activated cell sorting data were analyzed using FlowJo, version 10 (TreeStar Inc).
Immunoblot experiments
Cells were lysed and ran on a 15% acrylamide gel at 110 volts, and blotted overnight at 120 mA at 4°C on a polyvinylidene fluoride membrane. Membranes were incubated with primary anti-human antibodies in Tris-buffered saline with 0.5% Tween and 6% bovine serum albumin overnight at 4°C and a secondary anti-mouse antibody (Ab) at room temperature for 2 hours.
Coimmunoprecipitation experiments
Immunoprecipitation was performed by lysing cells in immunoprecipitation buffer (50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, protease inhibitors from Sigma-Aldrich). The lysates were precleared with Protein G–Sepharose Beads (GE Healthcare) and the tagged RhoG protein purified using anti-hemagglutinin agarose beads (Sigma-Aldrich). Immunoprecipitates were washed twice with immunoprecipitation buffer and once with wash buffer (50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid pH 8.0, 300 mM NaCl, 5 mM EDTA, 1% NP-40, 50 mM NaF, 1 mM Na3VO4, protease inhibitors; Sigma-Aldrich), and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis separation, blotting, and immunostaining using antibodies against RhoG, Munc13-4, glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz Biotechnology), and hemagglutinin tag (1:500 dilution for Munc13-4 antibody and 1:1000 dilution for others; Sigma-Aldrich).
Flow cytometry-based T- and NK-cell degranulation
T- and NK-cell degranulation were assessed by CD107a surface staining. For primary NK cells or NK-92 cells, K562 cells were used as targets. For CD8+ T cells, P815 target cells were preincubated either with or without anti-CD3 Ab (OKT-3, 0.1 µg/mL) and incubated with effector cells at 1:1 ratio for 5 hours. Following incubation, cells were stained with anti-CD3, CD57, and CD107a antibodies for 30 minutes, then subsequently washed and analyzed using LSR-Fortessa flow cytometer. For NK-92 experiments, effector cells were labeled with V450 proliferation dye and mixed with target cells at a 1:2 ratio. Cells were briefly spun down and incubated in the presence of anti-CD107a antibody for 4 hours.
T- and NK-cell cytotoxicity
Primary NK-cell cytotoxicity was measured using 51Cr-release assay as previously described.22 The cytotoxic activity of CD8+ T cells was assessed using flow cytometry-based assay with GFP-expressing P815 target cells coated with anti-CD3 Ab as previously described.23 The cytotoxic activity of NK-92 cells was assessed similarly, using GFP-expressing K562 target cells.
Phospholipid dot blot (lipid-binding assay)
Dot-blot experiments were carried out as described previously, with some modifications.24 Strips were incubated 30 minutes in Tris-buffered saline with Tween 20 (TBST; 0.15 M NaCl, 10 mM Tris-HCl, 0.05% Tween 20, pH 8.0) with 5% bovine serum albumin at room temperature and subsequently transferred to the purified tagged protein solution at 0.5 μg/mL in TBST overnight at 4°C. Each strip was then washed 3 times in TBST buffer before incubating with anti-His antibody (Santa Cruz Biotechnology) and a secondary horseradish peroxidase conjugate antibody solution. Antibody binding was detected using ECL Select (Amersham Biosciences).
Results
Identification of germline-encoded RhoG deficiency
We investigated a male patient born to healthy parents, who at the age of 4 months developed severe HLH. The disease presentation included characteristic HLH features, such as hemophagocytosis, hepatosplenomegaly, fever, cytopenias, low hemoglobin, hypertriglyceridemia, and elevated ferritin and soluble interleukin-2 receptor (Figure 1A-C; supplemental Figure 1H; supplemental Table 1, available on the Blood Web site). A detailed case report is provided in the supplemental Information. Because 8 of 8 diagnostic criteria for HLH were fulfilled, the patient was treated according to the HLH-2004 protocol.25 Targeted sequencing did not identify any germline mutations in established FHL-associated genes,2 and normal expression of these genes at protein level in patient-derived T and NK cells excluded potential effects of noncoding variants (supplemental Figure 1A-B). We hence performed whole-exome sequencing to identify a potential novel genetic etiology of HLH and found compound heterozygous mutations in the RHOG gene: a missense mutation (c.511G>A; p.Glu171Lys) and a 33-kb deletion (Chr11:3848730-3881730) (supplemental Data; supplemental Tables 2 and 3), respectively. The mode of inheritance of compound heterozygous RHOG mutations among family members was most consistent with an autosomal-recessive disorder. A quantitative polymerase chain reaction (qPCR) assay confirmed a reduced copy number of the RHOG gene in genomic DNA from the patient and his father (0.53- and 0.62-fold, respectively) carrying the monoallelic RHOG deletion (I-2, II-1; supplemental Figure 1C). The mother and a sibling were heterozygous for the RHOG missense mutation. The maternally inherited RHOG c.511G>A missense variant has not been observed in the 1000 Genomes Project and the Exome Aggregation Consortium database or Genome Aggregation Database and was predicted damaging with a combined annotation dependent depletion score of 31. There are 52 missense variants and 2 loss of function variants in the RHOG gene listed in the Genome Aggregation Database v2.1.1. database. All except 1 missense variant (p.Ala45Thr) are very rare, and only 1 missense variant (p.Ile137Val), which is predicted benign, is present in the database as a homozygote. The RhoG p.E171K mutation is located in the C-terminal α-helix of the GTPase domain, substituting a highly evolutionary conserved residue (Figure 1D-E). Immunoblotting analysis showed undetectable levels of protein in both patient T cells and fibroblasts (Figure 1F). Furthermore, upon ectopic expression of wild-type (WT) and mutant RHOG constructs in HEK293T cells, we observed significantly reduced amounts of mutant RhoG (Figure 1G), confirming the deleterious effect of the mutation on RhoG protein stability. To contextualize this novel gene defect and further support causality, we used a protein–protein interaction network analysis26 to quantitatively determine the degree of connectivity between the candidate gene identified in the patient and known disease-causing genes associated with exocytosis. Although RhoG functions have not been reported to be related to CG exocytosis so far, our protein–protein interaction network analysis (supplemental Figure 2A-B; supplemental Table 5) defined RhoG as the top candidate, most closely connected to gene products relevant to exocytosis. Additional analysis of the connectivity between human GTPases and disease-causing genes associated with an impaired cytotoxic function of human lymphocytes also identified RHOG as one of the top candidates among 139 small GTPase genes (supplemental Figure 2C). Collectively, our genetic and systems-level analyses strongly indicated a potential role of RhoG in exocytosis and loss of RhoG function as a novel etiology of HLH.
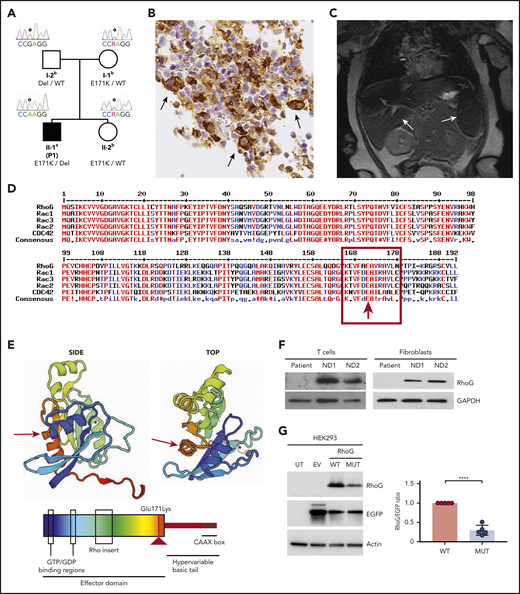
Identification of a genetic variant causing loss of RHOG expression. (A) Pedigree of the HLH patient family. Solid symbol (▪) indicates affected patient (P1); open symbol (○) corresponds to unaffected family members. The RHOG c.511G>A, p.Glu171Lys variant (RHOG gene reference sequences Ensembl: ENSG00000177105, ENST00000533217, GRCh37.p13) is indicated by E171K, the RHOG ∼33kb deletion is indicated as Del, the WT alleles by WT. aIndividual evaluated by whole-exome sequencing, Sanger sequencing, single nucleotide polymorphism (SNP) genotyping, and qPCR analysis. bIndividuals evaluated by targeted screening of the RHOG c.511G>A, p.Glu171Lys variant by Sanger sequencing, SNP genotyping, and qPCR analysis. (B) Immunohistochemistry of the patient bone marrow biopsy showing staining for CD68. Arrows indicate histiocytic cells with hemophagocytosis. (C) Abdominal magnetic resonance image revealing hepatosplenomegaly and abnormal signal intensity in both spleen and liver. (D) Amino acid alignment of the selected ρ family GTPase members demonstrating high conservation of the mutated residue. (E) A 3-dimensional model (top) and a scheme (bottom) of RhoG domain organization and localization of the missense mutation (red arrow). (F) Immunoblot analysis of the lysates from the patient and normal donor (ND) expanded T cells and fibroblasts. (G) Immunoblot analysis of the lysates from HEK293 cells untransfected (UT) and transfected with empty vector (EV) or vectors encoding WT and Glu171Lys mutant (MUT) RHOG genes. The difference in WT and mutated RhoG expression levels were quantified by normalization to EGFP expression (graph on right). Data are presented as the mean ± standard error of the mean (SEM). P values were calculated using a multiple Student t test. *****P < .0001.
Identification of a genetic variant causing loss of RHOG expression. (A) Pedigree of the HLH patient family. Solid symbol (▪) indicates affected patient (P1); open symbol (○) corresponds to unaffected family members. The RHOG c.511G>A, p.Glu171Lys variant (RHOG gene reference sequences Ensembl: ENSG00000177105, ENST00000533217, GRCh37.p13) is indicated by E171K, the RHOG ∼33kb deletion is indicated as Del, the WT alleles by WT. aIndividual evaluated by whole-exome sequencing, Sanger sequencing, single nucleotide polymorphism (SNP) genotyping, and qPCR analysis. bIndividuals evaluated by targeted screening of the RHOG c.511G>A, p.Glu171Lys variant by Sanger sequencing, SNP genotyping, and qPCR analysis. (B) Immunohistochemistry of the patient bone marrow biopsy showing staining for CD68. Arrows indicate histiocytic cells with hemophagocytosis. (C) Abdominal magnetic resonance image revealing hepatosplenomegaly and abnormal signal intensity in both spleen and liver. (D) Amino acid alignment of the selected ρ family GTPase members demonstrating high conservation of the mutated residue. (E) A 3-dimensional model (top) and a scheme (bottom) of RhoG domain organization and localization of the missense mutation (red arrow). (F) Immunoblot analysis of the lysates from the patient and normal donor (ND) expanded T cells and fibroblasts. (G) Immunoblot analysis of the lysates from HEK293 cells untransfected (UT) and transfected with empty vector (EV) or vectors encoding WT and Glu171Lys mutant (MUT) RHOG genes. The difference in WT and mutated RhoG expression levels were quantified by normalization to EGFP expression (graph on right). Data are presented as the mean ± standard error of the mean (SEM). P values were calculated using a multiple Student t test. *****P < .0001.
RhoG deficiency specifically impairs NK and CD8+ T-cell cytotoxicity
The key pathogenetic mechanism of classical HLH is defective lymphocyte-mediated killing, which results in hyperactivation of immune cells.2 Therefore, we assessed killing and degranulation efficiency in patient-derived primary NK cells and ex vivo expanded CD8+ T cells. These experiments revealed severely reduced killing activity and degranulation in patient NK and T cells (Figure 2A-C). Functional ex vivo assessment of the patient-derived T cells was not possible because of the extremely low peripheral blood numbers of cytotoxic CD8+CD57+ T cells (supplemental Figure 1I-J). To further evaluate how RHOG depletion affects the cytotoxic activity of human lymphocytes, we performed RHOG knock out (KO) in NK-92 cell line using CRISPR/Cas9 technology and small interfering RNA (siRNA)-based knockdown in primary human CD8+ T cells from healthy volunteers (Figure 2D-E). In both models, RHOG ablation caused reduction of cytotoxic activity without altering the expression levels of other HLH-associated proteins (supplemental Figure 1A,G). Furthermore, pharmacological inhibition of RhoG with ITX3, a small-molecule compound that blocks the RhoG guanine exchange factor Trio,27 significantly reduced the killing efficiency of primary CTLs and NK cells obtained from a healthy donor (Figure 2F). Finally, supplementation of RHOG KO NK-92 cells with a RHOG WT construct restored expression and degranulation (Figure 2G). Together, these data identify RhoG as an essential component of the exocytosis machinery in human NK and CD8+ T cells and provide proof of causality for RhoG deficiency as a novel subtype of HLH.
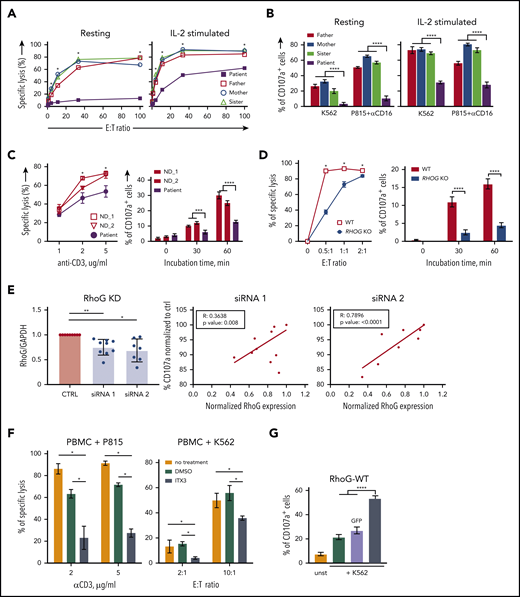
RhoG deficiency impairs NK and CD8+ T-cell cytotoxicity. (A) Target cell killing by primary NK cells from patient and healthy relatives upon coculture with K562 target cells. (B) Degranulation by primary NK cells from patient and healthy relatives assessed by CD107a surface exposure. (C) Target cell killing and degranulation in expanded CD8+ T cells from patient and normal donors upon coculture with P815 target cells coated with anti-CD3 mAb. (D) Killing and degranulation by NK-92 WT and RHOG KO cells upon coculture with K562 target cells for 24 hours. (E) siRNA-mediated RHOG KD in CD8+ T cells of healthy individuals. The efficiency of 2 siRNAs (#1 and #2) targeting RHOG compared with the control siRNA was calculated as a ratio between RhoG and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein levels (left). The effect of RHOG KD using 2 different siRNAs on degranulation of normal donor-derived CD8+ T cells shown as correlation between RhoG protein levels and degranulation efficiency (middle and right). (F) Effect of ITX3, an inhibitor of Trio-dependent RhoG activation, on killing capacity of primary T and NK cells. ND peripheral blood mononuclear cells (PBMC) were preincubated with ITX3 for 1 hour before the addition of target cells. Anti-CD3 monoclonal Ab-coated P815, and K562 cells were used to trigger T- and NK-cell cytotoxicity, respectively. (G) Degranulation assay showing reconstitution of impaired cytotoxic granules release in RHOG KO NK-92 cells upon transfection with plasmids encoding WT RHOG or GFP. All data are presented as the mean ± standard error of the mean (SEM). P values were calculated using a multiple Student t test and 2-way analysis of variance. *P < .05; **P < .01; ***P < .001; *****P < .0001.
RhoG deficiency impairs NK and CD8+ T-cell cytotoxicity. (A) Target cell killing by primary NK cells from patient and healthy relatives upon coculture with K562 target cells. (B) Degranulation by primary NK cells from patient and healthy relatives assessed by CD107a surface exposure. (C) Target cell killing and degranulation in expanded CD8+ T cells from patient and normal donors upon coculture with P815 target cells coated with anti-CD3 mAb. (D) Killing and degranulation by NK-92 WT and RHOG KO cells upon coculture with K562 target cells for 24 hours. (E) siRNA-mediated RHOG KD in CD8+ T cells of healthy individuals. The efficiency of 2 siRNAs (#1 and #2) targeting RHOG compared with the control siRNA was calculated as a ratio between RhoG and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein levels (left). The effect of RHOG KD using 2 different siRNAs on degranulation of normal donor-derived CD8+ T cells shown as correlation between RhoG protein levels and degranulation efficiency (middle and right). (F) Effect of ITX3, an inhibitor of Trio-dependent RhoG activation, on killing capacity of primary T and NK cells. ND peripheral blood mononuclear cells (PBMC) were preincubated with ITX3 for 1 hour before the addition of target cells. Anti-CD3 monoclonal Ab-coated P815, and K562 cells were used to trigger T- and NK-cell cytotoxicity, respectively. (G) Degranulation assay showing reconstitution of impaired cytotoxic granules release in RHOG KO NK-92 cells upon transfection with plasmids encoding WT RHOG or GFP. All data are presented as the mean ± standard error of the mean (SEM). P values were calculated using a multiple Student t test and 2-way analysis of variance. *P < .05; **P < .01; ***P < .001; *****P < .0001.
Because RhoG is reported to be involved in modulating the T-cell receptor signaling pathway,28 we hypothesized that RhoG loss of function might compromise lymphocyte activation. However, no defects in activation or proliferation of patient-derived T cells or RHOG KO NK-92 cells were detected (Figure 3A-D; supplemental Figure 3A-E). Consistent with observations in other forms of FHL, we observed markedly increased levels of interferon-γ (IFN-γ), a cytokine with a key pathogenic role in the disease, and CXCL9 in the plasma of the index patient (Figure 3E-F). Correspondingly, we found that production and release of IFN-γ by RhoG-deficient lymphocytes was unimpaired (Figure 3G-H; supplemental Figure 3G). Even upon suboptimal and physiological stimulation, we observed normal to slightly increased activation and IFN-γ production by RhoG-deficient NK and T cells (supplemental Figure 3H-J). Thus, our data indicate that RhoG deficiency specifically abrogates lymphocyte exocytosis without impairing the signaling downstream of activatory receptors and cytokine production.
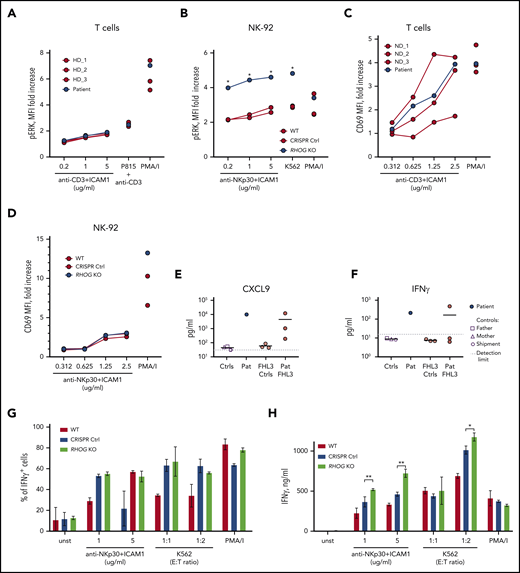
RhoG depletion does not affect lymphocyte activation nor cytokine production. (A) Graph comparing phosphorylated ERK1/2 in T cells upon stimulation with anti-CD3/hICAM1, anti-CD3–coated P815 cells, and PMA/I. Cells were analyzed by flow cytometry after intracellular staining with antibody recognizing phosphorylated ERK1/2 (Thr202/Tyr204). (B) ERK1/2 phosphorylation in WT and RHOG KO NK-92 cells upon stimulation. Analysis was performed similar to panel A. (C-D) Upregulation of CD69 on the surface of NK-92 and T cells upon stimulation with plate-bound Abs (anti-NKp30 or anti-CD3) and hrICAM1. The concentration of the titrated Ab is indicated on the x-axis. (E-F) Graphs show pathologically high levels of IFN-γ (E) and CXCL9 (IFN-γ–inducible chemokine) (F) in the plasma samples of the index patient (Pat) and healthy controls (father, mother, and shipment control, as indicated on the figure legend). Three samples from unrelated FHL3 patients (Pat FHL3) with active disease and corresponding controls (FHL3 Ctrl) were used for comparison. (G) Production of IFN-γ by WT and RHOG KO NK-92 cells upon stimulation with anti-NKp30+hrICAM1, K562 cells, and PMA/I. Cells were analyzed by flow cytometry after intracellular staining for IFN-γ. (H) Release of IFN-γ by WT and RHOG KO NK-92 cells measured 24 hours after stimulation by enzyme-linked immunosorbent assay in the culture medium. Data shown are representative of 2 independent experiments. P values were calculated using a multiple Student t test. *P < .05; **P < .01.
RhoG depletion does not affect lymphocyte activation nor cytokine production. (A) Graph comparing phosphorylated ERK1/2 in T cells upon stimulation with anti-CD3/hICAM1, anti-CD3–coated P815 cells, and PMA/I. Cells were analyzed by flow cytometry after intracellular staining with antibody recognizing phosphorylated ERK1/2 (Thr202/Tyr204). (B) ERK1/2 phosphorylation in WT and RHOG KO NK-92 cells upon stimulation. Analysis was performed similar to panel A. (C-D) Upregulation of CD69 on the surface of NK-92 and T cells upon stimulation with plate-bound Abs (anti-NKp30 or anti-CD3) and hrICAM1. The concentration of the titrated Ab is indicated on the x-axis. (E-F) Graphs show pathologically high levels of IFN-γ (E) and CXCL9 (IFN-γ–inducible chemokine) (F) in the plasma samples of the index patient (Pat) and healthy controls (father, mother, and shipment control, as indicated on the figure legend). Three samples from unrelated FHL3 patients (Pat FHL3) with active disease and corresponding controls (FHL3 Ctrl) were used for comparison. (G) Production of IFN-γ by WT and RHOG KO NK-92 cells upon stimulation with anti-NKp30+hrICAM1, K562 cells, and PMA/I. Cells were analyzed by flow cytometry after intracellular staining for IFN-γ. (H) Release of IFN-γ by WT and RHOG KO NK-92 cells measured 24 hours after stimulation by enzyme-linked immunosorbent assay in the culture medium. Data shown are representative of 2 independent experiments. P values were calculated using a multiple Student t test. *P < .05; **P < .01.
RhoG controls synaptic actin reorganization in human lymphocytes
RhoG has been shown to control various cellular functions through the regulation of cytoskeletal rearrangements.29-31 In lymphocytes, the active form of RhoG promotes actin polymerization and induces changes in cell morphology.32 Thus, we assessed motility and IS assembly in RhoG-deficient cytotoxic lymphocytes. Although only RhoG-deficient NK cells showed abnormalities in migratory capacity, both NK cells and T cells displayed cytoskeletal and morphological defects (supplemental Figure 4A-I). These morphological defects were associated with an overall decrease in filamentous actin (F-actin) levels (Figure 4A), but intact F-actin polarization toward the IS (supplemental Figure 5A). Moreover, the formation of stable conjugates by RHOG KO NK-92 cells with K562 target cells was normal (supplemental Figure 4I), and we did not observe striking abnormalities in the IS formed by these cells. Because RhoG has been shown to regulate actin cytoskeleton rearrangements through Rac1 and Cdc42,33,34 we assumed that the actin-related defects observed in RhoG-deficient lymphocytes might stem from the perturbed activity of the ρ GTPases. To test this assumption, we compared the activity levels of major actin cytoskeleton-regulating ρ GTPases: Rac1, RhoA, and Cdc42 in WT and RHOG KO NK-92 cells. Indeed, we noted a significant decrease in Rac1 and RhoA activation upon stimulation of NK-92 RHOG KO cells (Figure 4B; supplemental Figure 5B).
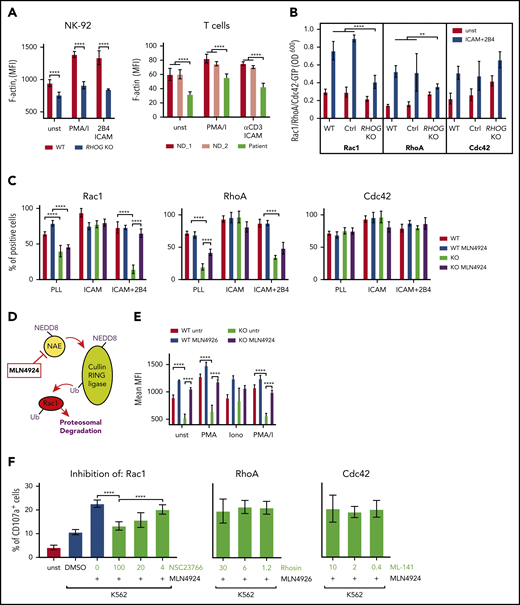
RhoG controls F-actin through modulation of Rac1 activity. (A) Graphs comparing F-actin intensity (phalloidin staining) in WT and RhoG-deficient NK-92 (left) and patient-derived T cells (right). (B) Histogram showing activation of Rac1, RhoA, and Cdc42 in WT and RHOG KO NK-92 cells upon stimulation. Activation of GTPases was measured using G-LISA assay (Cytoskeleton, Inc). (C) Histogram comparing activation of Rac1, RhoA, and Cdc42 in WT and RHOG KO NK-92 cells upon treatment with MLN4924. (D) Scheme demonstrating the mechanism of action for the top hit MLN4924 identified in pharmacological screening experiment. (E) Graph showing F-actin intensity in WT and RhoG-deficient NK-92 cells without, and after 16 hours of treatment with MLN4924. (F) Histogram demonstrating the effect of Rac1, RhoA, and Cdc42 inhibitors on degranulation in MLN4924-treated RHOG KO NK-92 cells. ICAM, human recombinant ICAM-1 Fc chimera; PLL, poly-L lysine. Data shown as mean ± SEM. P values were calculated using a multiple Student t test. **P < .01; *****P < .0001.
RhoG controls F-actin through modulation of Rac1 activity. (A) Graphs comparing F-actin intensity (phalloidin staining) in WT and RhoG-deficient NK-92 (left) and patient-derived T cells (right). (B) Histogram showing activation of Rac1, RhoA, and Cdc42 in WT and RHOG KO NK-92 cells upon stimulation. Activation of GTPases was measured using G-LISA assay (Cytoskeleton, Inc). (C) Histogram comparing activation of Rac1, RhoA, and Cdc42 in WT and RHOG KO NK-92 cells upon treatment with MLN4924. (D) Scheme demonstrating the mechanism of action for the top hit MLN4924 identified in pharmacological screening experiment. (E) Graph showing F-actin intensity in WT and RhoG-deficient NK-92 cells without, and after 16 hours of treatment with MLN4924. (F) Histogram demonstrating the effect of Rac1, RhoA, and Cdc42 inhibitors on degranulation in MLN4924-treated RHOG KO NK-92 cells. ICAM, human recombinant ICAM-1 Fc chimera; PLL, poly-L lysine. Data shown as mean ± SEM. P values were calculated using a multiple Student t test. **P < .01; *****P < .0001.
The activity of Rac1 is crucial for the synaptic F-actin remodeling35 and has a strong impact on CG release by human lymphocytes.36,37 To understand whether RhoG deficiency causes a defect in degranulation through impaired Rac1 signaling and associated actin remodeling, we looked for the strategies to specifically manipulate Rac1 activity in the RhoG-defective background. Pharmacological screening has been successfully used to explore the druggability of small GTPases and to dissect associated pathways.38,39 Hence, we used compounds from an anticancer drug collection library and identified the NEDD8-activating enzyme inhibitor MLN4924 as a small-molecule compound able to rescue the impaired Rac1 activation in RhoG-deficient NK-92 cells (Figure 4C; supplemental Figure 5B-C). This finding is in line with Rac1 being a substrate of cullin-RING ligases controlled by the NEDD8 pathway.40,41 Inhibition of this pathway by MLN4924 blocks the degradation of Rac1, resulting in its accumulation, which may augment actin cytoskeletal dynamics (Figure 4D). Indeed, the drug-mediated rescue of active Rac1 levels led to a full correction of the actin cytoskeleton organization defects characterized in RHOG KO NK-92 cells (Figure 4E), thereby demonstrating that the control of actin remodeling by RhoG is exerted via Rac1. Furthermore, using Rac1, RhoA, and Cdc42 inhibitors, we confirmed that Rac1 activity is specifically required for MLN4924-mediated rescue of degranulation (Figure 4F). More detailed exploration of the effect of RhoG deficiency on the synaptic F-actin meshwork by airy-scan microscopy revealed that although actin density was globally decreased at the IS of RHOG KO NK-92 cells, actin clearance zones (local areas free from F-actin within the IS) were reduced (Figure 5A-C). This suggests that, in the absence of RhoG, the cortical actin meshwork undergoes restructuring that may block CG access to the PM. We also observed the impaired formation of F-actin foci in IS of RHOG KO NK-92 cells. Diminished dense F-actin foci were also detected in the CG neighborhood (Figure 5D-F). Surprisingly, these striking defects in synaptic actin were almost entirely rescued by MLN4924 treatment. Our assumption was that the rescue of Rac1 activity and associated actin remodeling would lead to a normalization of the degranulation activity of the RhoG-deficient NK-92 cells. However, we observed only a moderate effect of MLN4924 on amelioration of exocytosis in these cells (supplemental Figure 5C-D). Comparably, forced Rac1 expression restored F-actin organization in RHOG KO NK-92 cells, but did only partially restore the degranulation phenotype (Figure 5G-H; supplemental Figure 5C-D). Importantly, MLN4924 had a comparable effect on patient-derived T cells (supplemental Figure 5E-F). Together, our data imply that the Rac1-dependent cytoskeletal defects in RhoG-deficient lymphocytes might contribute, but do not play a dominant role in the observed degranulation phenotype. Consequently, RhoG may be endowed with a yet uncharacterized, noncanonical function to control CG exocytosis in cytotoxic lymphocytes.
![RhoG regulates synaptic F-actin density and architecture in human lymphocytes. (A) Representative images demonstrating synaptic F-actin network (gray, upper) in WT and RHOG KO NK-92 cells with or without MLN4924 treatment. Lower panel shows corresponding images from upper panel after convolution. (B) Graph showing percentage of surface (area) within the IS covered by F-actin. (C) Comparison of the F-actin density (brightness) in the IS of WT and RHOG KO NK-92 cells with or without MLN4924 treatment. (D) Representative images demonstrating colocalization of synaptic F-actin meshwork (red) and CG (green) in NK-92 cells. White frames show magnified F-actin neighborhood around individual CG. (E) Graph indicating difference (shown as standard deviation [SD]) of F-actin density around individual CGs. (F) Scheme illustrating the effect of RhoG-deficiency and MLN4924 treatment on the of F-actin density in CG neighborhood. Reconstitution of the F-actin density (brightness) (G) and degranulation capacity (H) in WT and RHOG KO NK-92 cells upon supplementation with constitutively active (Q61L) RAC1 and RHOG constructs. Scale bars, 5 μm. Data on the graphs shown as mean ± SEM. P values were calculated using Student t test. ***P < .001; *****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020008738/1/m_bloodbld2020008738f5.png?Expires=1770510687&Signature=cn9KSNOlfTl1fOIgbtNuJ6-V-cnpNhsu0UgWkx2~Svv~bCHv4GctuMm8hkGyU0rS4QDZKRxTZHy4-PlSNp72Wb~W-vtrZlhdSQ6v2kvM5PXn9UoRGLsNFdO76vilRJWJBavAiL6JAygD5L1~UhpIVrM04by-MNY3W2OzqdRIsU-W7fLxKVSKBHyy4tekLP-0NUePDI0~qdRSsnrgW2c3ZxtOG-kDzP8bfnRijO7LG0JoXn8Pzf-job682sl8tsLmxf4RPNH2RVznPWeMwQbeEcDhxssFU5OYhm7EYYVWM2ZTysoXjYNNUWhxSPgNs5gzPruv3neVPQc05-emKEW85w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
RhoG regulates synaptic F-actin density and architecture in human lymphocytes. (A) Representative images demonstrating synaptic F-actin network (gray, upper) in WT and RHOG KO NK-92 cells with or without MLN4924 treatment. Lower panel shows corresponding images from upper panel after convolution. (B) Graph showing percentage of surface (area) within the IS covered by F-actin. (C) Comparison of the F-actin density (brightness) in the IS of WT and RHOG KO NK-92 cells with or without MLN4924 treatment. (D) Representative images demonstrating colocalization of synaptic F-actin meshwork (red) and CG (green) in NK-92 cells. White frames show magnified F-actin neighborhood around individual CG. (E) Graph indicating difference (shown as standard deviation [SD]) of F-actin density around individual CGs. (F) Scheme illustrating the effect of RhoG-deficiency and MLN4924 treatment on the of F-actin density in CG neighborhood. Reconstitution of the F-actin density (brightness) (G) and degranulation capacity (H) in WT and RHOG KO NK-92 cells upon supplementation with constitutively active (Q61L) RAC1 and RHOG constructs. Scale bars, 5 μm. Data on the graphs shown as mean ± SEM. P values were calculated using Student t test. ***P < .001; *****P < .0001.
RhoG regulates synaptic F-actin density and architecture in human lymphocytes. (A) Representative images demonstrating synaptic F-actin network (gray, upper) in WT and RHOG KO NK-92 cells with or without MLN4924 treatment. Lower panel shows corresponding images from upper panel after convolution. (B) Graph showing percentage of surface (area) within the IS covered by F-actin. (C) Comparison of the F-actin density (brightness) in the IS of WT and RHOG KO NK-92 cells with or without MLN4924 treatment. (D) Representative images demonstrating colocalization of synaptic F-actin meshwork (red) and CG (green) in NK-92 cells. White frames show magnified F-actin neighborhood around individual CG. (E) Graph indicating difference (shown as standard deviation [SD]) of F-actin density around individual CGs. (F) Scheme illustrating the effect of RhoG-deficiency and MLN4924 treatment on the of F-actin density in CG neighborhood. Reconstitution of the F-actin density (brightness) (G) and degranulation capacity (H) in WT and RHOG KO NK-92 cells upon supplementation with constitutively active (Q61L) RAC1 and RHOG constructs. Scale bars, 5 μm. Data on the graphs shown as mean ± SEM. P values were calculated using Student t test. ***P < .001; *****P < .0001.
RhoG regulates exocytosis through interaction with Munc13-4 and CG docking to the plasma membrane
No obvious molecular link between human RhoG and the exocytotic machinery has been reported to date. To explore the possibility for such a link, we performed affinity purification of Strep-hemagglutinin-tagged RhoG with subsequent mass spectrometry analysis of isolated complexes from human lymphocytes. Among the identified RhoG protein partners, we found only 1 protein directly associated with exocytosis: Munc13-4 (Figure 6A; supplemental Table 7). In humans, this protein is encoded by the UNC13D gene. Mutations in UNC13D have been described to selectively disrupt the exocytotic release of CGs by cytotoxic lymphocytes, causing classical FHL3.7 Biochemical pulldown experiments from the lysates of Jurkat and NK-92 cells confirmed the interaction between RhoG and Munc13-4, and implied that this interaction is activation dependent (Figure 6B; supplemental Figure 6A). Additional cell microscopy-based studies revealed colocalization of endogenous Munc13-4 with ectopically expressed RhoG WT or constitutively active RhoG (Q61L), but not with dominant-negative (N17T) RhoG mutants (Figure 6C; supplemental Figure 6B). These data suggest that RhoG may facilitate the process of exocytosis through its interaction with Munc13-4 upon lymphocyte stimulation.
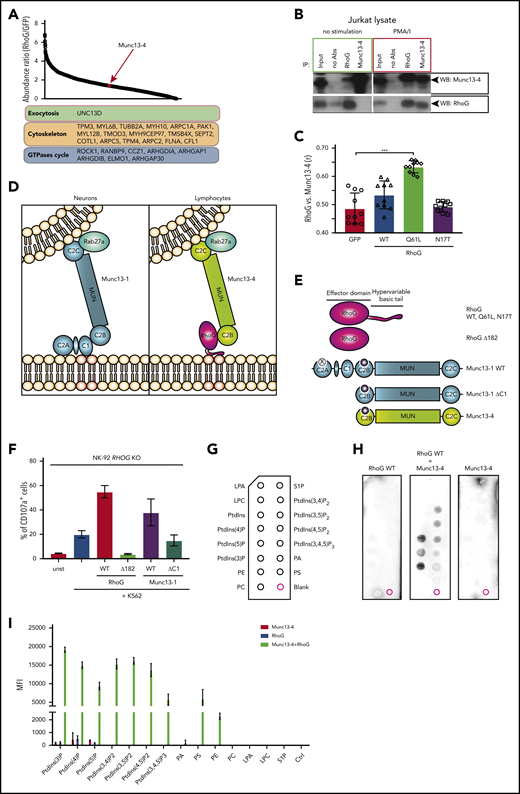
RhoG facilitates CG docking through interaction with Munc13-4 and cooperative binding to plasma membrane lipids. (A) Affinity purification of STREP-HA-tagged RhoG protein complexes identified novel interacting protein partners of RhoG, including Munc13-4. (B) Coimmunoprecipitation of endogenous RhoG with Munc13-4 from a lysate of unstimulated or phorbol 12-myristate 13-acetate + ionomycin (PMA/I)-stimulated Jurkat cells. (C) Quantification of colocalization of RhoG and Munc13-4 in Jurkat cells (representative microscopy images are shown in supplemental Figure 4F). (D) A proposed model of RhoG-mediated exocytosis specific for human lymphocytes. (E) RHOG and UNC13D constructs used in reconstitution experiments and lipid-binding assay. (F) Graph demonstrating reconstitution of degranulation in RHOG KO NK-92 cells with constructs encoding WT and truncated (Δ182) RhoG, WT, and truncated (ΔC1) Munc13-1. (G) Scheme of PIP-strip membrane depicting spots of lipid species. (H) PIP-strip assay with recombinant WT RhoG protein alone or in a mixture with purified Munc13-4, showing binding pattern to the lipids indicated in panel G. (I) Quantification of PIP-strip assay results, showing a lipid-binding pattern of recombinant proteins.
RhoG facilitates CG docking through interaction with Munc13-4 and cooperative binding to plasma membrane lipids. (A) Affinity purification of STREP-HA-tagged RhoG protein complexes identified novel interacting protein partners of RhoG, including Munc13-4. (B) Coimmunoprecipitation of endogenous RhoG with Munc13-4 from a lysate of unstimulated or phorbol 12-myristate 13-acetate + ionomycin (PMA/I)-stimulated Jurkat cells. (C) Quantification of colocalization of RhoG and Munc13-4 in Jurkat cells (representative microscopy images are shown in supplemental Figure 4F). (D) A proposed model of RhoG-mediated exocytosis specific for human lymphocytes. (E) RHOG and UNC13D constructs used in reconstitution experiments and lipid-binding assay. (F) Graph demonstrating reconstitution of degranulation in RHOG KO NK-92 cells with constructs encoding WT and truncated (Δ182) RhoG, WT, and truncated (ΔC1) Munc13-1. (G) Scheme of PIP-strip membrane depicting spots of lipid species. (H) PIP-strip assay with recombinant WT RhoG protein alone or in a mixture with purified Munc13-4, showing binding pattern to the lipids indicated in panel G. (I) Quantification of PIP-strip assay results, showing a lipid-binding pattern of recombinant proteins.
Having established the interaction between RhoG and Munc13-4, we aimed to further investigate the molecular mechanism through which this interaction might regulate CG exocytosis in human lymphocytes. RhoG can assist the guanine nucleotide exchange factor Trio in binding to membrane phospholipids.24 The basic C-terminal membrane-binding tail of RhoG controls Trio translocation and attachment to the PM through cooperative binding to membrane lipids such as phosphatidylinositol 3,4-bisphosphate (PtdIns[3,4]P2). Therefore, RhoG can serve as a “molecular glue” to anchor molecular partners to the PM. Munc13-4 is predominately expressed in human bone marrow and lymphoid tissues, whereas Munc13-1, Munc13-2, and Munc13-3 are preferentially expressed in neurons. Unlike neuronal Munc13s, Munc13-4 lacks an N-terminal C1 lipid-binding domain.42 We conjectured that RhoG might use its membrane-binding capacity to dock Munc13-4–coated CGs to the PM of lymphocytes, which is a critical step during CG exocytosis. Using total internal reflection fluorescence microscopy, we confirmed the increased number of motile CG in the IS of RHOG KO NK-92 cells suggestive of a docking defect, reported previously for UNC13D-deficient cells (supplemental Figure 6C). We propose that this mechanism is tissue-specific and restricted to hematopoietic cells, congruent with the clinical phenotype of the patient that did not display overt central nervous system abnormalities. Indeed, using neuronal Munc13-1 constructs (Figure 6D-E) as experimental tools to test our hypothesis, we found that overexpression of neuronal full-length Munc13-1, but not Munc13-1 lacking its C1 membrane-binding domain (Munc13-1ΔC1), rescued CG exocytosis in RHOG KO NK-92 cells (Figure 6F). This result strongly suggests that the degranulation defect observed in RhoG-deficient lymphocytes is due to inability of Munc13-4 to bind the PM. To further challenge our hypothesis, we decided to ablate membrane-binding properties of RhoG by generating a truncated RhoG construct lacking the C-terminal membrane-binding tail (RhoGΔ182, residues 1-182). Unlike RhoG WT, the RhoGΔ182 construct did not rescue CG exocytosis in RHOG KO NK-92 cells (Figure 6F). Next, we directly assessed the ability of RhoG to target Munc13-4 to membrane lipids. We produced recombinant His6-tagged WT or mutant RhoG proteins as well as Munc13-4 in bacteria and used mixtures of the purified proteins in a protein-lipid overlay assay (PIP-strip assay). We found that RhoG is capable of binding phospholipids in an activation-dependent manner, and although Munc13-4 alone is not binding to lipids, a mixture of RhoG and Munc13-4 interacts with phospholipids (Figure 6G-I; supplemental Figure 6D).
These experiments showed that the requirement for RhoG in human lymphocyte exocytosis is based on the previously unrecognized ability of RhoG to attach Munc13-4+ CGs to the PM. This, in turn, enables subsequent CG tethering and fusion with the PM, facilitating the release of CG cargo toward the target cell.
Discussion
Familial or primary HLH denotes a group of disorders characterized by severe systemic hyperinflammation and requires prompt diagnosis and treatment initiation (reviewed in Canna and Marsh43 ). The understanding of molecular pathomechanisms of HLH might have a significant effect on disease prognosis and management.44 The typical primary or familiar forms of HLH (FHL types 2-5) are caused by the defects in PRF1, UNC13D, STX11, and STXBP2 genes and result in specific abrogation of perforin-mediated killing by CTL and NK cells. Other HLH-associated syndromes and actin-related immune disorders are often associated with the additional phenotypic features.11,12,43 In our study, we have discovered a novel, nonconventional role of the small GTPase RhoG in the regulation of lymphocyte cytotoxicity. At the core of this function is the direct control of CG exocytosis that links RhoG to the immunopathology of HLH. RhoG has been previously associated with the regulation of microtubule and actin cytoskeleton dynamics.31,45-47 Notably, a role in regulating exocytosis machinery has been proposed for some actin regulating small GTPases such as Rac1 and Cdc42. In MIN6 β cells (a model cell line for pancreatic β cells), Cdc42 interacts with vesicle-associated membrane protein 2 to induce the release of insulin-containing vesicles.48 In neuroendocrine cells, Rac1 has been shown to activate phospholipase D1, which generates phosphatidic acid on the PM to promote its fusion with vesicles.49 In human lymphocytes, Rac1 and Cdc42 are also contributing to exocytosis, but rather through the regulation of cortical actin, and their direct role in the control of the exocytosis machinery is not defined yet. Given distinct mechanisms and unique interaction partners linking Rac1, RhoG, and Cdc42 to exocytosis, one could speculate that the precise role of these GTPases in exocytosis is cell- or tissue-specific. A recent study50 revealed the landscape of interactions for 28 ρ family GTPases in HEK and HeLa cells using proximity-dependent biotinylation. Although this work indeed suggests a certain level of cell type-specific differences in the GTPases’ interactome, it does not cover this issue in detail. Our model of RhoG-dependent exocytosis mediated through the interaction with Munc13-4 provides an explanation for tissue specificity of exocytosis defect and clinical phenotype of RhoG deficiency. Despite the important role of RhoG in neurons48 and other cell types, our index patient presented with a classical FHL, while no overt other syndromic disease features, such as developmental defects, have been registered before or after hematopoietic stem cell transplantation. We explain that RhoG deficiency affects primarily immune cells by its interaction with hematopoietic Munc13-4, which in contrast to other Munc13 isoforms lack the C1 lipid-binding domain51 and thus require RhoG assistance for binding to membrane lipids.
In our biochemical experiments, we showed that RhoG affinity to the membrane lipids drastically increases upon binding to Munc13-4. Previously, cooperative binding to phospholipids has been shown for the Trio-RhoG complex.24 Interestingly, Munc13-4-RhoG and Trio-RhoG complexes display distinct lipid specificity. Such preferential binding to distinct phospholipids may contribute to the spatial coordination of RhoG functional activities with respect to exocytosis vs cytoskeletal regulation. Recent studies show how dynamic changes in lipid composition contribute to F-actin reorganization across the synapse.52,53 Effector molecules that stabilize and deconstruct synaptic F-actin require distinct localization within the IS. On the other hand, colocalization of factors disrupting F-actin with components of exocytosis machinery would greatly facilitate the delivery and release of CGs. Given the ability of RhoG to form complexes with both F-actin and exocytosis regulators, it would allow it to locally regulate the relocalization of these molecules within the IS. The mechanisms of RhoG-mediated exocytosis discovered in our work show how lipid-mediated signaling may coordinate actin cytoskeleton remodeling and activation of the exocytosis machinery, 2 key events essential for the cytotoxic function of human lymphocytes.
The molecular and cellular signatures of classical FHL are distinct from other HLH-associated syndromic disorders. In a recent study, de novo mutations in CDC42 have been associated with the development of HLH as part of an autoinflammatory condition called NOCARH (neonatal onset of pancytopenia, autoinflammation, rash, and episodes of HLH) syndrome.54 This work revealed that the reduction of cytotoxic activity in lymphocytes with Cdc42 dysfunction is a result of an actin-related defect that compromises conjugate formation and impairs migration. This is in contrast to RhoG deficiency in which ablation of cytotoxic functions in T and NK cells is directly caused by the defect in the exocytosis machinery. Importantly, aberrant Cdc42 function results in additional clinical features, such as dyshematopoiesis. This was not the case for our RhoG-deficient patient, and Cdc42 activity was normal in our cellular models. We propose that biallelic RHOG mutations abrogating its expression specifically impair exocytosis in cytotoxic lymphocytes and ultimately result in FHL. Based on the current FHL classification,25 RHOG mutations may tentatively represent FHL type 6; however, future studies will need to confirm this phenotype in additional patients. The spectrum of this disease may possibly be broader than HLH because of the documented role of RhoG in other cell types, including neutrophils,55 platelets,56 and macrophages.57 Munc13-4 is also critical for exocytosis in several of these hematopoietic cell types, yet clinically autosomal recessive UNC13D mutations phenocopy PRF1 mutations causative of FHL3 and FHL2, respectively. Further studies will delineate the clinical spectrum of RhoG deficiency. Given the important role of lymphocyte cytotoxicity for the elimination of tumor cells, one may speculate that similar to other HLH-related hypomorphic mutations,3-5 reduced functionality of RhoG (eg, via hypomorphic mutations or haploinsufficiency of RHOG) may be associated with increased cancer susceptibility.
In summary, our study uncovers a novel mechanism of exocytosis regulated by the small ρ family GTPase RhoG, providing insights into how exocytosis is regulated in different types of hematopoietic cells. Our findings establish an essential role of RhoG in human lymphocyte exocytosis. We speculate that this mechanism is especially suited to the rapidly forming transient synapses of the immune system, dynamically coordinating actin reorganization and vesicle docking. These insights offer new avenues for future studies of multilayered regulatory mechanisms coordinating exocytosis in non-neuronal cells.
For original data, please contact the corresponding author (kaan.boztug@ccri.at).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patient’s family for participating in the study and all the members of the research groups and clinicians for the discussions. The authors also thank Sophie Allart and Astrid Canivet-Laffite for their help and support at the Cell Imaging Facility of the Center of Pathophysiology of Toulouse Purpan, Toulouse, France, and Marion Gröger and Sabine Rauscher from the Imaging Core Facility of the Medical University of Vienna (Vienna, Austria) for their assistance. In addition, the authors thank the Sequencing Unit of Institute for Molecular Medicine Finland Technology Centre for NGS Library preparation and sequencing.
This work was supported by the European Research Council through ERC Consolidator Grant 820074 (K.B.), the Vienna Science and Technology Fund through Project LS14-031 (K.B.), the Austrian Academy of Science through DOC Fellowships 25365 (J.H.) and 25225 (M. Thian), the Finnish Foundation for Pediatric Research and Pediatric Research Center, Helsinki University Hospital (M.R.J.S.), and the Swedish Research Council, Cancer Foundation, Children's Cancer Foundation, and the Knut and Alice Wallenberg Foundation (Y.T.B.).
Authorship
Contribution: A.K., L.D., K.B., M.R.J.S., J.S., and Y.T.B. initiated the study; A.K., G.P.C., J.H., S.C.C., D.G., B.H., Y.G., M. Thian, J.P., and Ö.Y.P. performed the experiments; J.S. designed the study for the genetic analysis; L.T. conducted exome sequencing, statistical analyses, and validation of the genetic findings; T.T. provided material from the patients and clinical information, conducted the medical follow-up, and provided expertise in critical reading; M. Taskinen, S.K., A.H., O.L., and K.V. provided clinical information and expertise; M.R.J.S., M.J.K., and A.J.L. critically reviewed the manuscript and provided expertise and feedback; A.K., A.J.L., K.B., Y.T.B., and M.R.J.S. wrote the manuscript with additional input from the other authors; K.B., Y.T.B., and J.S. secured and provided funding for the experiments outlined; K.B. coordinated the study with input from Y.T.B., M.R.J.S., and J.S.; and all authors critically reviewed the manuscript and agreed to its publication.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kaan Boztug, St Anna Children’s Cancer Research Institute (CCRI) and Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases (LBI-RUD), Zimmermannplatz 10, A-1090 Vienna, Austria; e-mail: kaan.boztug@ccri.at.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal