

In this issue of Blood, 1 identify hematoxylin as an inhibitor of the glycan binding domain of calreticulin (CALR).
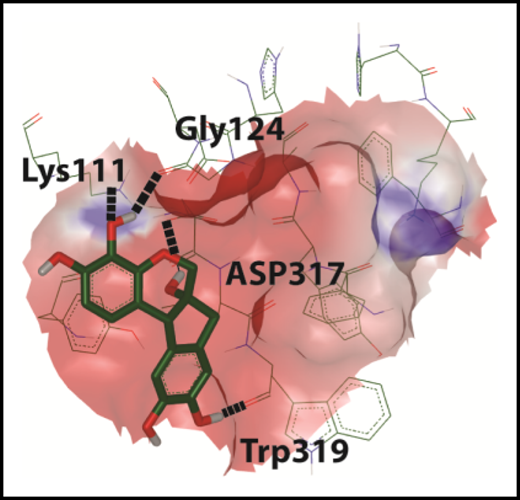
In silico docking of hematoxylin onto the glycan binding domain of CALR.9 See Figure 2B in the article by Jia et al that begins on page 1920.
In silico docking of hematoxylin onto the glycan binding domain of CALR.9 See Figure 2B in the article by Jia et al that begins on page 1920.
The discovery of CALR mutations in myeloproliferative neoplasms (MPNs) filled in a missing piece of the pathobiological puzzle for these disorders.2,3 Mutations in Janus activated kinase 2 and the thrombopoietin receptor (MPL) had been previously identified as important molecular drivers of disease, but they did not explain all classical MPN cases.4 Mutations in CALR were identified in a large fraction of these remaining cases,2,3 but their biological role in driving myeloproliferation was not immediately apparent.
CALR is an endoplasmic reticulin (ER)-resident protein with multifaceted biological roles, including mediating proper protein folding and regulating calcium homeostasis. CALR has a lectin (glycan binding) domain, which allows it to serve as a chaperone by interacting with glycans on nascently synthesized proteins. MPN-associated insertions or deletions in the CALR coding region cause a +1-bp frameshift, which alters the amino acid sequence of the C terminus.3 This changes a negatively charge C terminus to one enriched in positively charged amino acids. This frameshift also abolishes an ER-retrieval motif, which would normally enable CALR to be retrieved from the Golgi back to the ER.5 In the absence of this motif, CALR fails to maintain its ER residency and travels toward the cell surface.
How do these changes to the protein sequence promote a myeloproliferative disorder? Expression of mutant CALR in murine models leads to thrombocytosis.6,7 CALR mutations confer cytokine independence and cellular transformation in a MPL-dependent manner.6-8 Frameshift mutations alter the conformation of CALR to enable robust interaction of its lectin domain with MPL.8 This stabilizes MPL as an dimer,9 activating the receptor to enhance downstream signaling and drive myeloproliferation.
Jia et al set out to precisely target this molecular interaction. To do this, they harnessed existing crystal structures of CALR to make an in silico model of the glycan binding domain (see figure). They used this model for molecular docking studies to predict drugs that might interact with the glycan binding domain. Intriguingly, one of these identified molecules, hematoxylin, selectively inhibits the growth of CALR-mutant–expressing BaF3 cells. Hematoxylin is long familiar to hematologists as a dye used in hematoxylin and eosin staining. When used as a dye, hematoxylin is further modified by oxidation and complexed with metals. Hematoxylin, as used in these studies, is the unmodified form. As an extension of these findings, they show that other compounds in the same class as hematoxylin (catechols) also show selectivity toward CALR-mutant cells, although none quite as impressive as hematoxylin itself.
In a coculture model, CALR wild-type cells outcompeted CALR-mutant cells in the presence of hematoxylin. Hematoxylin also induces the apoptosis of CALR-mutant cells. To evaluate whether these effects of hematoxylin are indeed due to interaction with the CALR glycan binding domain, the authors created substitutions at critical residues in this domain. These glycan binding domain mutations abrogate the effects of hematoxylin. Finally, they show that hematoxylin reduces the colony-forming unit potential of CALR-mutant human leukemia samples in culture.
At the molecular level, Jia et al demonstrate using a bioluminescence resonance energy transfer assay that hematoxylin inhibits the interaction of mutant CALR with MPL. This is consistent with idea that binding of hematoxylin to the lectin domain disrupts its ability to interact with MPL’s glycans. Furthermore, the disruption of this interaction dampens activation of STAT5 downstream of MPL. Together, these data show that small-molecule binding to the lectin domain of CALR can prevent it from interacting with MPL and thus block aberrant MPL activation.
Glycans are often underappreciated players in biology, but nevertheless, they make up a substantial portion of the structure of membrane proteins. Glycans dictate membrane protein folding, confer structure, and mediate interactions with other molecules. The story of MPL and mutant CALR elegantly highlights the importance of glycans and their interacting lectins. Lectins have been targeted in myeloid malignancy before. Gemtuzumab ozogamicin, a drug developed for acute myeloid leukemia, targets the sialic acid binding lectin CD33.10 However, the specific disruption of lectin binding to its target to hamper malignant function is an exciting next step in the modulation of glycan-protein interactions for therapeutic benefit.
This study provides us with an important proof of principle that the glycan binding domain of calreticulin is targetable. With micromolar potency and as-yet-unclear in vivo tolerability, hematoxylin itself likely serves as a starting point for further drug development. However, with these studies, mutant calreticulin has revealed itself as vulnerable, and that is something celebrate.
Conflict-of-interest disclosure: J.E.M. receives research funding from the Gilead Research Scholars Award Program.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal