

Introduction:
CalDAG-GEFI (Calcium and diacyglycerol-regulated guanine exchange factor I) deficiency or platelet-type bleeding disorder-18 (BDPLT18) is a relatively new, rare hemorrhagic disorder with autosomal recessive inheritance (OMIM #615888). CalDAG-GEFI activates Rap1 and is essential for αIIbβ3 (GP IIb/IIIa) integrin activation in platelets, a key step in platelet aggregation. The deficiency is due to mutations of the encoding gene, RASGRP2 (HGNC ID 9879), which is located on chromosome 11q13.1. To date, 23 pathogenic variants in the RASGRP2 gene are listed in the Human Gene Mutation Database (HGMD®). Two other RASGRP2 splice-site mutations (c.74-1G>C and 372-3C>G) are described in the literature in patients suffering from CalDAG-GEFI loss of function.
Patients and Methods:
We investigated a 3 yr-old index patient who was referred to our outpatient clinic because of Glanzmann thrombasthenia (GT)-like symptoms and GT-typical pattern in platelet aggregometry analyses. The boy presented with hematomas and recurrent infections, postnatally, he had shown petechiae. After a tongue bite at the age of 1 yr, he needed tranexamic acid to stop bleeding. So far, no surgery has been performed. His family members including mother, father, elder sisters and brother had no history of bleeding. Informed consent for our study of biochemical and molecular genetic characterization for inherited platelet disorders using NGS was given by all family members.
Platelet functions were assessed by light transmission aggregometry using citrated PRP. Membrane glycoprotein quantification (CD41, CD42a and CD42b), fibrinogen binding, von Willebrand factor (VWF) binding and the investigation of alpha- and delta- granule secretion were performed by flow cytometry. For the index patient and his parents we performed NGS panel sequencing (Nextera Rapid Custom Enrichment, Illumina). 80 genes known to be associated with platelet disorders were screened. The variants detected were classified based on the 2015 American College of Medical Genetics (ACMG) guidelines. ClinVar, Human Gene Mutation Database, and in silico pathogenicity prediction were used to assess the detected variants. The segregation of the putative mutation was confirmed by Sanger sequencing of three unaffected siblings.
Results:
The index patient presented with normal values for platelet count, aPTT, INR, FXIII and VWF parameters. ADP (4, 10 and 20µM/L), epinephrine (8, 16 and 40µM/L) and low concentration collagen (2µM/mL) and TRAP6 (5µM/mL) induced aggregation was markedly reduced. However, normal values after stimulation with 10µM/mL collagen and TRAP6 respectively were achieved. Agglutination after stimulation with ristocetin (1,2mg/mL) was normal. Flow cytometry revealed severely decreased fibrinogen binding after stimulation with ADP (0.25-5.0 μmol/l) compared to control platelets. For CD41 (GP IIb/IIIa-complex), CD42a/b (GP Ib/IX-complex), ristocetin-induced VWF-binding, CD62- and CD63-expression, normal values were measured.
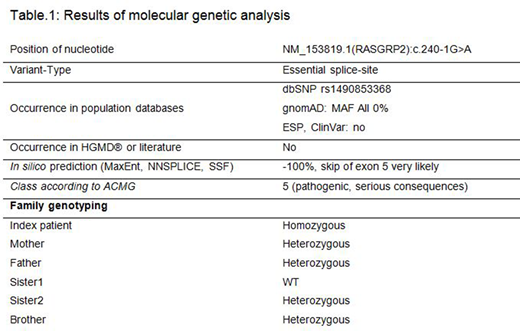
Molecular genetic analysis revealed a novel homozygous splice-site mutation of the RASGRP2 gene in the index patient. The mutation was heterozygous in the non- consanguineous parents (Tab.1).
Conclusion:
We have identified a novel recessive mutation of the RASGRP2 gene in a boy affected by a Glanzmann thrombasthenia-like disorder. Platelet aggregometry analysis showed typical failure of aggregation with low dose collagen and TRAP6, whereas aggregation after stimulation with higher concentration reached normal values. Flow cytometric measured severely reduced fibrinogen binding indicated a functional defect of the αIIbβ3 fibrinogen receptor with normal expression. Comprehensive platelet function analysis and panel sequencing allowed early and precise diagnosis and therapy of this ultra-rare platelet signaling defect.
Zieger:Biotest: Research Funding; DFG: Research Funding; CSL Behring: Research Funding; Takeda: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal