

Key Points
SEC22B interacts with a region of NBEAL2 where 2 missense variants associated with GPS have been identified.
Knockout of SEC22B or NBEAL2 expression in megakaryocytic cells results in failure of α-granule biogenesis.

Abstract
Studies of inherited platelet disorders have provided many insights into platelet development and function. Loss of function of neurobeachin-like 2 (NBEAL2) causes gray platelet syndrome (GPS), where the absence of platelet α-granules indicates NBEAL2 is required for their production by precursor megakaryocytes. The endoplasmic reticulum is a dynamic network that interacts with numerous intracellular vesicles and organelles and plays key roles in their development. The megakaryocyte endoplasmic reticulum is extensive, and in this study we investigated its role in the biogenesis of α-granules by focusing on the membrane-resident trafficking protein SEC22B. Coimmunoprecipitation (co-IP) experiments using tagged proteins expressed in human HEK293 and megakaryocytic immortalized megakaryocyte progenitor (imMKCL) cells established binding of NBEAL2 with SEC22B, and demonstrated that NBEAL2 can simultaneously bind SEC22B and P-selectin. NBEAL2-SEC22B binding was also observed for endogenous proteins in human megakaryocytes using co-IP, and immunofluorescence microscopy detected substantial overlap. SEC22B binding was localized to a region of NBEAL2 spanning amino acids 1798 to 1903, where 2 GPS-associated missense variants have been reported: E1833K and R1839C. NBEAL2 containing either variant did not bind SEC22B coexpressed in HEK293 cells. CRISPR/Cas9-mediated knockout of SEC22B in imMKCL cells resulted in decreased NBEAL2, but not vice versa. Loss of either SEC22B or NBEAL2 expression resulted in failure of α-granule production and reduced granule proteins in imMKCL cells. We conclude that SEC22B is required for α-granule biogenesis in megakaryocytes, and that interactions with SEC22B and P-selectin facilitate the essential role of NBEAL2 in granule development and cargo stability.
Introduction
Human platelets are small anucleate cells derived from megakaryocytes (MKs) resident in the bone marrow. Platelets initiate hemostasis by responding to vessel injury and triggering the formation of a fibrin clot.1 Beyond hemostasis, platelets are also implicated in physiological processes that include innate immunity, host defense, wound healing, and tissue regeneration.2-5 They are also involved in pathological conditions such as malignancy, atherosclerosis, and thrombotic diseases including acute coronary syndrome and stroke.6-9 MKs release platelet precursors (ie, proplatelets) and platelets into the sinusoidal vasculature via processes that are under active investigation. Aspects identified so far include microtubule-dependent proplatelet extension,10-12 MK protrusion driven by membrane dynamics,13 and rapid MK fragmentation and platelet release (ie, MK rupture) in response to an acute increase in platelet demand.14
Key to the functions of platelets is their ability to secrete a wide range of small molecules and proteins carried in dense (δ)- and α-granules, respectively. These granules are classed as lysosome-related organelles,15-19 and insights into their production by MKs have come from investigating hereditary disorders where they are deficient.18,20 α-granule deficiencies include arthrogryposis, renal dysfunction, and cholestasis (ARC) syndrome (Online Mendelian Inheritance in Man [OMIM]: 608552 and 613401) and gray platelet syndrome (GPS; OMIM: 614169), where VPS33B, VPS16B (VIPAS39), and NBEAL2 have been identified as proteins required for α-granule biogenesis.21-25 NBEAL2 is a large (>300 kDa) protein with putative functional regions that include the BEACH domain,26-28 which is shared by a family of proteins thought to serve as intracellular scaffolds.26 Progress in understanding the role of NBEAL2 in α-granule biogenesis has been modest, hampered in part by the fact that unlike VPS33B24 and VPS16B,25 its expression is largely limited to hematopoietic cells.29
Studies of platelet granule deficiencies using patient material and an assortment of mouse and cellular models have yielded insights into cellular mechanisms of granule biogenesis.27,30-33 Key observations include: (1) loss of δ-granules does not compromise the formation of α-granules and vice versa, indicating distinct developmental pathways24,25,27,32,34 ; (2) loss of VPS33B and/or VPS16B results in the absence of α-granules and their distinctive membrane protein P-selectin in platelets, whereas loss of NBEAL2 results in loss of α-granules but not P-selectin,18,24,25,27,32,35 indicating that VPS33B/VPS16B act upstream of NBEAL2 in α-granule biogenesis; (3) lack of NBEAL2 causes MK-derived and endocytosed α-granule cargo proteins to be externalized via RAB11-associated late recycling endosomes, indicating that NBEAL2 is required for α-granule cargo loading and/or retention27,35 ; and (4) NBEAL2 associates with P-selectin in MKs.35
Recently, novel insights have been reported regarding the role of the endoplasmic reticulum (ER), an elaborate and dynamic network of membrane cisternae and tubules that extends throughout cells and interacts with a wide range of organelles, including mitochondria, peroxisomes, the Golgi apparatus, endosomes and the plasma membrane.36-39 Many ER-organelle interactions are facilitated by membrane contact sites, where membranes are closely apposed and tethered via protein complexes that influence organelle dynamics.37,39 Flexible ER tubules are involved in organelle trafficking, fusion, and fission events,39 all of which have been linked to endosome maturation, leading to the expectation that the ER likely plays a central role in α-granule production by MKs. Studies using methods such as affinity purification–mass spectrometry, coimmunoprecipitation (co-IP), and BioID proximity biotinylation assays have identified several potential protein interactors for VPS33B31,40,41 and NBEAL2.35,42 SEC22B, an ER-resident SNARE (soluble N-ethylmaleimide–sensitive fusion protein attachment protein receptor),43,44 has been reported to interact with VPS33B.31,41 Some ER-resident proteins are concentrated in a particular region; for example, the chaperone calnexin is largely present in the protein-synthetic ER, and we have previously observed little overlap of calnexin with NBEAL2.35 SEC22B, in contrast, is present throughout the ER, and in preliminary immunofluorescence microscopy studies we observed considerable overlap with NBEAL2 in primary human MKs. In this study, we examined the potential role of SEC22B and SEC22B-NBEAL2 interactions in α-granule development.
We initiated our studies using tagged proteins expressed in HEK293 cells, where we detected binding of SEC22B with NBEAL2 via co-IP experiments. This interaction was confirmed for endogenous SEC22B and NBEAL2 in primary human MKs, where we also observed intracellular colocalization of SEC22B and NBEAL2 via immunofluorescence microscopy. Building on our earlier demonstration that NBEAL2 binds P-selectin, we observed that it can simultaneously bind SEC22B. We mapped the SEC22B binding region of NBEAL2 to a location between amino acids (aa) 1798 and 1903, a region where 2 GPS-causing missense variants have been reported: E1833K and R1839C.22 NBEAL2 constructs containing either E1833K or R1839C failed to bind SEC22B, suggesting a link between NBEAL2-SEC22B binding and α-granule production. We used CRISPR/Cas9 gene editing to knock out SEC22B and NBEAL2 in respective lines of megakaryocytic immortalized MK progenitor (imMKCL) cells and observed that the loss of either protein resulted in decreased α-granule proteins and the absence of α-granules identified via electron microscopy. Expression of tagged versions of SEC22B or NBEAL2 in knockout (KO) imMKCL cells restored α-granule protein expression. Therefore, both SEC22B and NBEAL2 are required for MK α-granule production, where their interaction may play a key role.
Methods
Plasmid construct generation, IP, antibodies, immunofluorescence microscopy, immunoblot analysis of protein content, and statistical analysis are described in the supplemental Methods, available on the Blood Web site.
Human MK culture
Human MKs were cultured as previously described.45 Details are provided in supplemental Methods.
Culture of imMKCL cells and generation of KO and rescue lines
Megakaryocytic imMKCL cells were cultured as previously described.46 Details are provided in supplemental Methods.
Immunofluorescence microscopy
Preparation and immunostaining of primary human MKs were performed as previously described.45 Confocal laser fluorescence images were obtained with oil immersion objectives (60×/1.35 NA) using a Quorum spinning disc confocal inverted fluorescence microscope as described47 and were analyzed and prepared with Volocity (Perkin-Elmer) and Adobe Photoshop. Details are provided in supplemental Methods.
Electron microscopy
Transmission electron microscopy of imMKCL cells was performed as previously described.27 Briefly, cells were fixed with 2.5% glutaraldehyde in 0.1 M of cacodylate at pH of 7.4, postfixed with 2% osmium tetroxide in double-distilled water, dehydrated in acetone, and then embedded in Epon-Araldite. Thin sections were cut and stained with uranyl acetate and lead citrate, and samples were examined with a JEOL JEM-1011 electron microscope at 80 kV. Images were captured with a side-mounted Advantage HR CCD camera (Advanced Microscopy Techniques) and processed with Adobe Photoshop.
Results
NBEAL2 interacts with SEC22B in cell lysates
Interaction of SEC22B with NBEAL2 was examined via co-IP experiments performed with hemagglutinin (HA)-NBEAL2 and FLAG-SEC22B tagged proteins expressed in HEK293 cells. HA-NBEAL2 pulled down FLAG-SEC22B (Figure 1A), and FLAG-SEC22B pulled down HA-NBEAL2 (Figure 1B). In these experiments both proteins were overexpressed in HEK293 cells, which do not normally express significant amounts of NBEAL2. The potential of endogenously expressed NBEAL2 and SEC22B to associate was examined using human MK lysates, where SEC22B was able to coimmunoprecipitate NBEAL2 (Figure 1C), and NBEAL2 pulled down SEC22B (Figure 1D). These experiments confirmed the ability of NBEAL2 and SEC22B expressed by primary human MKs to interact at endogenous levels.
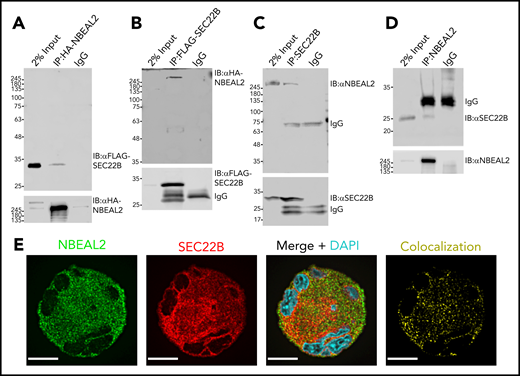
Interaction of tagged NBEAL2 and SEC22B in HEK293 cells, and native proteins in MKs. (A) HA-tagged protein was immunoprecipitated from lysates of HEK293 cells expressing both HA-tagged NBEAL2 (HA-NBEAL2) and FLAG-tagged SEC22B (FLAG-SEC22B). Immunoblots of lysate (input) and immunoprecipitates probed with anti-FLAG and anti-HA antibodies show HA-NBEAL2 pulled down FLAG-SEC22B. (B) IP of FLAG-tagged protein showed that FLAG-SEC22B pulled down HA-NBEAL2. (C) Lysate from primary human MKs was immunoprecipitated with anti-SEC22B antibody; immunoblot analysis shows that endogenously expressed SEC22B pulled down native NBEAL2. (D) IP of NBEAL2 from primary human MK lysate showed that NBEAL2 pulled down SEC22B. (E) Fixed primary human MKs immunostained for NBEAL2 (green) and SEC22B (red) were imaged via laser fluorescence confocal microscopy. XY profiles of midcell z sections from a representative mature cell show substantial overlap of NBEAL2 and SEC22B (bars = 10 μm). Images were obtained with an oil immersion objective (60×/1.35 NA) using a Quorum spinning disc confocal inverted fluorescence microscope. DAPI, 4′,6-diamidino-2-phenylindole; IgG, immunoglobulin G.
Interaction of tagged NBEAL2 and SEC22B in HEK293 cells, and native proteins in MKs. (A) HA-tagged protein was immunoprecipitated from lysates of HEK293 cells expressing both HA-tagged NBEAL2 (HA-NBEAL2) and FLAG-tagged SEC22B (FLAG-SEC22B). Immunoblots of lysate (input) and immunoprecipitates probed with anti-FLAG and anti-HA antibodies show HA-NBEAL2 pulled down FLAG-SEC22B. (B) IP of FLAG-tagged protein showed that FLAG-SEC22B pulled down HA-NBEAL2. (C) Lysate from primary human MKs was immunoprecipitated with anti-SEC22B antibody; immunoblot analysis shows that endogenously expressed SEC22B pulled down native NBEAL2. (D) IP of NBEAL2 from primary human MK lysate showed that NBEAL2 pulled down SEC22B. (E) Fixed primary human MKs immunostained for NBEAL2 (green) and SEC22B (red) were imaged via laser fluorescence confocal microscopy. XY profiles of midcell z sections from a representative mature cell show substantial overlap of NBEAL2 and SEC22B (bars = 10 μm). Images were obtained with an oil immersion objective (60×/1.35 NA) using a Quorum spinning disc confocal inverted fluorescence microscope. DAPI, 4′,6-diamidino-2-phenylindole; IgG, immunoglobulin G.
NBEAL2 and SEC22B overlap in the outer ER of human MKs
Confocal fluorescence microscopy imaging of immunostained fixed human MKs was used to examine the distributions of NBEAL2 and SEC22B, which showed substantial overlap (Figure 1E). In a previous study we observed that NBEAL2 overlaps extensively with P-selectin in α-granules/precursors, but not with calnexin, a chaperone localized primarily to the synthetic ER.35 The potential relationships of these proteins within human MKs was examined in cells stained for NBEAL2, SEC22B, and either calnexin (Figure 2A), P-selectin (Figure 2B), or the sarcoplasmic reticulum/ER calcium ATPase SERCA3 (Figure 2C; colocalization channels are shown in supplemental Figure 1). Images were used for a pairwise colocalization analysis based on Pearson’s correlation coefficient (Figure 2D; n = 20-62 cells). The results confirmed relatively strong overlap of NBEAL2 with both P-selectin and SEC22B, and a much weaker association with calnexin. SEC22B also showed a relatively strong association with P-selectin, supporting the possibility of a tripartite interaction of these proteins with NBEAL2. The overlap of SEC22B with calnexin seemed to be restricted to the region of the synthetic ER, and as expected SEC22B showed more extensive overlap with SERCA3 in the outer ER, as did NBEAL2.
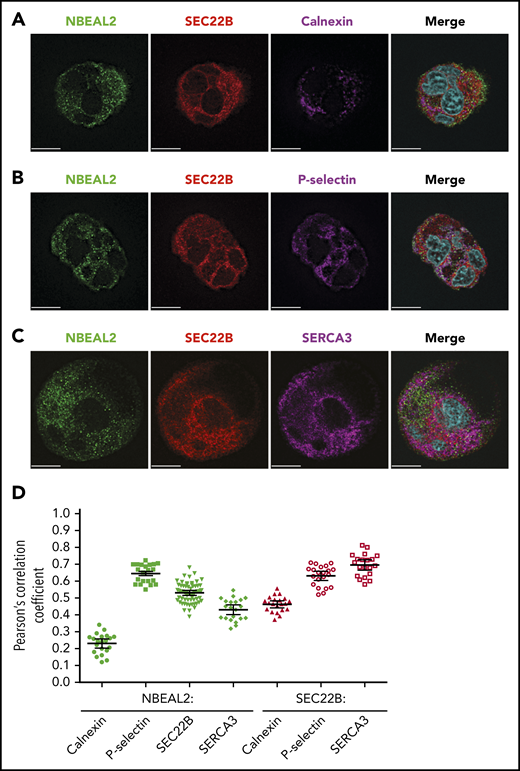
NBEAL2, SEC22B, and membrane-associated proteins in human MKs. Primary human MKs stained for NBEAL2 (green), SEC22B (red), and 1 of the following: calnexin (synthetic ER; magenta) (A), P-selectin (α-granules/precursors; magenta) (B), or SERCA3 (cortical ER; magenta; bars = 10 μm) (C). (D) Colocalization analysis based on Pearson’s correlation coefficient for NBEAL2-calnexin (n = 21 cells), NBEAL2–P-selectin (n = 21 cells), NBEAL2-SEC22B (n = 62 cells), NBEAL2-SERCA3 (n = 21 cells), SEC22B-calnexin (n = 21 cells), SEC22B–P-selectin (n = 21 cells), and SEC22B-SERCA3 (n = 20 cells; bars = 95% confidence intervals) indicates strong overlap among NBEAL2, SEC22B and P-selectin, with NBEAL2 having a weak association with calnexin. Images obtained as described in Figure 1. Colocalization channel images are shown in supplemental Figure 1.
NBEAL2, SEC22B, and membrane-associated proteins in human MKs. Primary human MKs stained for NBEAL2 (green), SEC22B (red), and 1 of the following: calnexin (synthetic ER; magenta) (A), P-selectin (α-granules/precursors; magenta) (B), or SERCA3 (cortical ER; magenta; bars = 10 μm) (C). (D) Colocalization analysis based on Pearson’s correlation coefficient for NBEAL2-calnexin (n = 21 cells), NBEAL2–P-selectin (n = 21 cells), NBEAL2-SEC22B (n = 62 cells), NBEAL2-SERCA3 (n = 21 cells), SEC22B-calnexin (n = 21 cells), SEC22B–P-selectin (n = 21 cells), and SEC22B-SERCA3 (n = 20 cells; bars = 95% confidence intervals) indicates strong overlap among NBEAL2, SEC22B and P-selectin, with NBEAL2 having a weak association with calnexin. Images obtained as described in Figure 1. Colocalization channel images are shown in supplemental Figure 1.
Defining the SEC22B binding region within NBEAL2
Regions within NBEAL2 involved with SEC22B interactions were identified via co-IP experiments in HEK293 cells (Figure 3). FLAG-tagged full-length SEC22B was coexpressed with HA-tagged full-length NBEAL2, or constructs expressing fragments containing various combinations of known functional domains (Figure 3A). As expected, HA-tagged full-length NBEAL2 pulled down FLAG-SEC22B (Figure 3B). Interaction with SEC22B was also observed with a construct containing the C-terminal PH-BEACH and WD40 domains (N2; aa 1798-2754), but not with the N-terminal region containing the armadillo-like and concanavalin A–like domains (N1; aa 1-1145; Figure 3B). The location of the SEC22B binding region was further refined using constructs encompassing the region preceding the NBEAL2 PH-BEACH domain, with the longer (N3; aa 1380-1903) being able to coimmunoprecipitate SEC22B, but not the shorter (N4; aa 1380-1797; Figure 3B). This result indicated that a SEC22B binding site is localized within aa 1798 to 1903 of NBEAL2. This was confirmed when a construct expressing this region (N5; aa 1798-1903) was able to pull down SEC22B (Figure 3B).
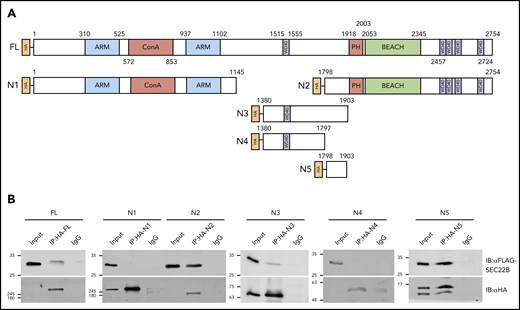
Identification of a SEC22B binding domain within NBEAL2. (A) HA-tagged full-length (FL) NBEAL2 and fragments N1 to N5 (initial and terminal amino acids indicated, plus known functional domains) were individually coexpressed with FLAG-SEC22B in HEK293 cells. (B) HA-tagged proteins were immunoprecipitated from lysates and analyzed via immunoblots. Probing with anti-FLAG and anti-HA showed that FLAG-SEC22B pulled down FL HA-NBEAL2, N2 (aa 1798-2754), N3 (aa 1380-1903), and N5 (aa 1798-1903), but not N1 (aa 1-1145) or N4 (aa 1380-1797). These results indicate that a region within aa 1798 to 1903 of NBEAL2 can bind SEC22B. ARM, armadillo-like; ConA, concanavalin A-like; IgG, immunoglobulin G.
Identification of a SEC22B binding domain within NBEAL2. (A) HA-tagged full-length (FL) NBEAL2 and fragments N1 to N5 (initial and terminal amino acids indicated, plus known functional domains) were individually coexpressed with FLAG-SEC22B in HEK293 cells. (B) HA-tagged proteins were immunoprecipitated from lysates and analyzed via immunoblots. Probing with anti-FLAG and anti-HA showed that FLAG-SEC22B pulled down FL HA-NBEAL2, N2 (aa 1798-2754), N3 (aa 1380-1903), and N5 (aa 1798-1903), but not N1 (aa 1-1145) or N4 (aa 1380-1797). These results indicate that a region within aa 1798 to 1903 of NBEAL2 can bind SEC22B. ARM, armadillo-like; ConA, concanavalin A-like; IgG, immunoglobulin G.
GPS-associated variants E1833K and R1839C abrogate NBEAL2-SEC22B binding
Two GPS-associated missense NBEAL2 variants, Glu1833Lys (E1833K) and Arg1839Cys (R1839C), have been reported within the 1798 to 1903 aa SEC22B-binding region (Figure 4A).22 To examine the potential effect of these variants on NBEAL2-SEC22B interaction, we coexpressed FLAG-tagged SEC22B with HA-tagged full-length NBEAL2 constructs containing the wild-type (WT) sequence or one of the variants in HEK293 cells. Co-IP experiments showed that WT NBEAL2 pulled down SEC22B as expected, whereas NBEAL2 containing the E1833K or R1839C variant was unable to coimmunoprecipitate SEC22B (Figure 4B). We previously demonstrated that WT NBEAL2 can coimmunoprecipitate P-selectin.35 The specificity of the effects of the NBEAL2 variants was examined via co-IP experiments that showed HA-tagged constructs expressing WT NBEAL2, E1833K, and R1839C all pulled down GFP-tagged P-selectin (Figure 4C). These results indicate specific effects of E1833K and R1839C on NBEAL2-SEC22B binding.
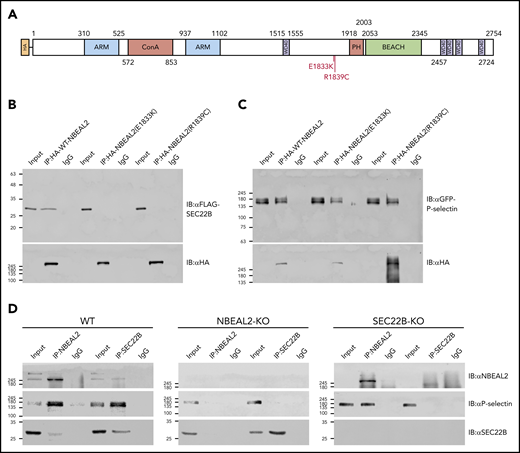
GPS-associated missense variants abrogate NBEAL2-SEC22B binding, and NBEAL2 is required for SEC22B–P-selectin interaction. (A) Two GPS-associated missense variants E1833K and R1839C are located within the putative SEC22B binding region of NBEAL2 (aa 1798-1903). (B) Transfected HEK293 cells coexpressing FLAG-SEC22B and either HA-tagged WT NBEAL2 (lanes 1-3), NBEAL2 (E1833K) (lanes 4-6), or NBEAL2 (R1839C) (lanes 7-9) were used for IP experiments and analyzed via immunoblots. Although all HA-tagged constructs were expressed, WT NBEAL2, but not the 2 NBEAL2 variants was able to coimmunoprecipitate SEC22B. (C) Similar experiments performed with HEK293 cells coexpressing GFP-tagged P-selectin instead of FLAG-SEC22B showed that WT NBEAL2 and both NBEAL2 variants were able to coimmunoprecipitate P-selectin, indicating a specific effect of GPS-associated variants on SEC22B binding. (D) Native NBEAL2 and SEC22B were immunoprecipitated using specific antibodies in WT, NBEAL2-KO, and SEC22B-KO imMKCL cells. NBEAL2, P-selectin, and SEC22B can be coimmunoprecipitated together in WT cells (WT, left panel). However, SEC22B was unable to coimmunoprecipitate P-selectin in the absence of NBEAL2 (NBEAL2-KO, middle panel), whereas NBEAL2 could coimmunoprecipitate P-selectin in the absence of SEC22B (SEC22B-KO, right panel). These results suggest that NBEAL2 mediates the interaction between SEC22B and P-selectin.
GPS-associated missense variants abrogate NBEAL2-SEC22B binding, and NBEAL2 is required for SEC22B–P-selectin interaction. (A) Two GPS-associated missense variants E1833K and R1839C are located within the putative SEC22B binding region of NBEAL2 (aa 1798-1903). (B) Transfected HEK293 cells coexpressing FLAG-SEC22B and either HA-tagged WT NBEAL2 (lanes 1-3), NBEAL2 (E1833K) (lanes 4-6), or NBEAL2 (R1839C) (lanes 7-9) were used for IP experiments and analyzed via immunoblots. Although all HA-tagged constructs were expressed, WT NBEAL2, but not the 2 NBEAL2 variants was able to coimmunoprecipitate SEC22B. (C) Similar experiments performed with HEK293 cells coexpressing GFP-tagged P-selectin instead of FLAG-SEC22B showed that WT NBEAL2 and both NBEAL2 variants were able to coimmunoprecipitate P-selectin, indicating a specific effect of GPS-associated variants on SEC22B binding. (D) Native NBEAL2 and SEC22B were immunoprecipitated using specific antibodies in WT, NBEAL2-KO, and SEC22B-KO imMKCL cells. NBEAL2, P-selectin, and SEC22B can be coimmunoprecipitated together in WT cells (WT, left panel). However, SEC22B was unable to coimmunoprecipitate P-selectin in the absence of NBEAL2 (NBEAL2-KO, middle panel), whereas NBEAL2 could coimmunoprecipitate P-selectin in the absence of SEC22B (SEC22B-KO, right panel). These results suggest that NBEAL2 mediates the interaction between SEC22B and P-selectin.
NBEAL2 can simultaneously bind SEC22B and P-selectin
We previously showed that NBEAL2 interacts with P-selectin in MKs and platelets,35 and we wanted to examine how SEC22B may fit into this interaction. We first observed that FLAG-SEC22B did not pull down GFP–P-selectin in lysate from HEK293 cells expressing both proteins (supplemental Figure 2 lanes 1-3), indicating they do not bind each other. When HA-NBEAL2 was expressed together with these constructs, FLAG-SEC22B was able to pull down both P-selectin and NBEAL2 (supplemental Figure 2 lanes 4-6). This indicates that NBEAL2 can bind both SEC22B and P-selectin simultaneously to form complexes containing at least 1 molecule of each. Similar experiments were performed in WT, NBEAL2-KO, and SEC22B-KO imMKCL cells, where native NBEAL2 and SEC22B were immunoprecipitated in each cell line using antibodies specific for these proteins (Figure 4D). In WT imMKCL cells, both NBEAL2 and SEC22B could coimmunoprecipitate P-selectin. Although NBEAL2 could coimmunoprecipitate P-selectin in the absence of SEC22B, SEC22B could not coimmunoprecipitate P-selectin in the absence of NBEAL2 (Figure 4D). These results suggest that NBEAL2 serves as a bridge in a tripartite interaction with P-selectin and SEC22B.
SEC22B KO in imMKCL cells results in loss of α-granules
We previously used CRISPR/Cas9-mediated gene editing and lentiviral transduction to generate imMKCL cells46 where ARPC1B expression was abrogated.47 A similar protocol was used to generate imMKCL KO cell lines lacking expression of NBEAL2 and SEC22B. Previous studies of imMKCL cells have demonstrated that mature cells are capable of forming α-granules detectable by transmission electron microscopy.48 We confirmed this observation in WT and mock lentiviral–transduced cells (Figure 5A-B), where α-granules were clearly visible. As expected from previous studies, α-granules were not observed in NBEAL2-KO cells (Figure 5C), and they also appeared to be absent in SEC22B-KO cells (Figure 5D). Additional electron micrographs are shown in supplemental Figure 4. Quantification of α-granules and mitochondria confirmed the absence of α-granules in NBEAL2-KO and SEC22B-KO imMKCL cells (supplemental Figure 5A), whereas the numbers of mitochondria were not significantly different (supplemental Figure 5B). These results confirm the expectation that NBEAL2 is required for α-granule formation in imMKCL cells, and they indicate that SEC22B is also required for this process.
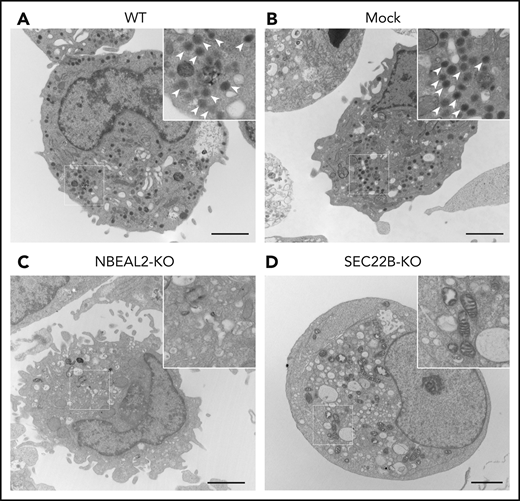
NBEAL2 and SEC22B are required for α-granule formation in imMKCL cells. Transmission electron micrographs of representative cells show abundant electron-dense α-granules (arrow heads in magnified insets) visible in WT (A) and empty lentiviral transduced (mock) (B) cells. Additional imMKCL cells are shown in supplemental Figure 4. α-granules were not observed in NBEAL2-KO (C) or SEC22B-KO (D) cells, indicating both proteins are required for granule formation (bars = 2 μm). Thin sections were cut and stained with uranyl acetate and lead citrate, and samples were examined with a JEOL JEM-1011 electron microscope at 80 kV. Images were captured with a side-mounted Advantage HR CCD camera.
NBEAL2 and SEC22B are required for α-granule formation in imMKCL cells. Transmission electron micrographs of representative cells show abundant electron-dense α-granules (arrow heads in magnified insets) visible in WT (A) and empty lentiviral transduced (mock) (B) cells. Additional imMKCL cells are shown in supplemental Figure 4. α-granules were not observed in NBEAL2-KO (C) or SEC22B-KO (D) cells, indicating both proteins are required for granule formation (bars = 2 μm). Thin sections were cut and stained with uranyl acetate and lead citrate, and samples were examined with a JEOL JEM-1011 electron microscope at 80 kV. Images were captured with a side-mounted Advantage HR CCD camera.
Loss of SEC22B does not cause major changes in the ER of imMKCL cells
The effects of SEC22B or NBEAL2 KO on the imMKCL cell ER were examined by assessing the intracellular distributions of ER-resident proteins via immunofluorescence microscopy (supplemental Figure 3). Comparisons with WT imMKCL cells indicated that SEC22B-KO and NBEAL2-KO cells did not show signs of gross changes in the distribution of either calnexin (supplemental Figure 3A) or SERCA3 (supplemental Figure 3B). As expected, SEC22B showed overlap with both calnexin and SERCA3 in WT and NBEAL2-KO cells. These results indicate that there are no major disruptions of the ER in imMKCL cells lacking either SEC22B or NBEAL2.
Loss of SEC22B or NBEAL2 reduces α-granule components in imMKCL cells
To rule out off-target effects of CRISPR/Cas9 gene editing, rescue lines were created by introducing expression of HA-tagged NBEAL2 or SEC22B, respectively, into NBEAL2-KO and SEC22B-KO cells to generate NBEAL2-KO-rescue (NBEAL2-R) and SEC22B-KO-rescue (SEC22B-R) cells (Figure 6A). Protein expression was analyzed via immunoblotting of cell lysates (Figure 6B) and densitometric analysis (Figure 6C). This analysis showed that SEC22B-KO cells had decreased expression of NBEAL2 (39.2% ± 11.8% relative to WT), which was rescued by expression of HA-SEC22B. In contrast, NBEAL2-KO, NBEAL2-R, and WT cells had similar levels of SEC22B (Figure 6C). Cells lacking either NBEAL2 or SEC22B showed marked decreases in von Willebrand factor, thrombospondin-1, and P-selectin (Figure 6C), indicating that loss of either protein negatively affects α-granule biogenesis.
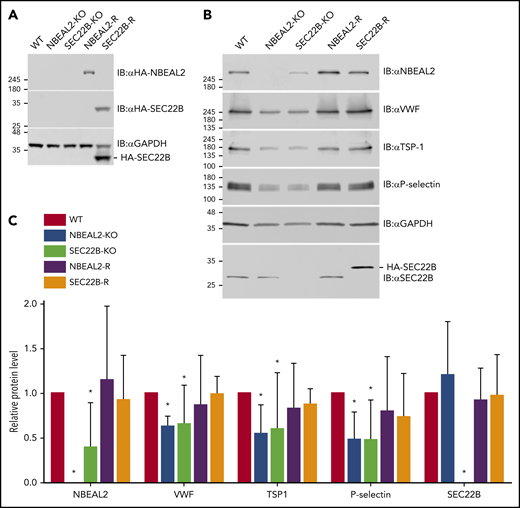
Expression of NBEAL2, SEC22B, and α-granule proteins in imMKCL WT, KO, and rescue cells. (A) Immunoblot analysis confirmed loss of NBEAL2 or SEC22B in CRISPR/Cas9 gene-edited NBEAL2-KO and SEC22B-KO cell lines and confirmed expression of HA-NBEAL2 and HA-SEC22B in the rescue lines NBEAL2-R and SEC22B-R. (B) Immunoblot analysis showed reduced expression of von Willebrand factor (VWF), thrombospondin-1 (TSP-1), and P-selectin in NBEAL2-KO and SEC22B-KO cells and restoration of expression of these proteins in NBEAL2-R and SEC22B-R cells. (C) Densitometric analysis of immunoblots indicated SEC22B-KO cells had significantly decreased NBEAL2 expression relative to WT (39.2% ± 11.8%), whereas NBEAL2-KO cells had normal levels of SEC22B (119.8% ± 14.0%). Relative protein levels represent the ratio relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) normalized to the WT ratio. Data are representative of 3 independent experiments (bars = 95% confidence intervals). *P < .05 via 2-way analysis of variance with Dunnett’s post hoc test relative to WT.
Expression of NBEAL2, SEC22B, and α-granule proteins in imMKCL WT, KO, and rescue cells. (A) Immunoblot analysis confirmed loss of NBEAL2 or SEC22B in CRISPR/Cas9 gene-edited NBEAL2-KO and SEC22B-KO cell lines and confirmed expression of HA-NBEAL2 and HA-SEC22B in the rescue lines NBEAL2-R and SEC22B-R. (B) Immunoblot analysis showed reduced expression of von Willebrand factor (VWF), thrombospondin-1 (TSP-1), and P-selectin in NBEAL2-KO and SEC22B-KO cells and restoration of expression of these proteins in NBEAL2-R and SEC22B-R cells. (C) Densitometric analysis of immunoblots indicated SEC22B-KO cells had significantly decreased NBEAL2 expression relative to WT (39.2% ± 11.8%), whereas NBEAL2-KO cells had normal levels of SEC22B (119.8% ± 14.0%). Relative protein levels represent the ratio relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) normalized to the WT ratio. Data are representative of 3 independent experiments (bars = 95% confidence intervals). *P < .05 via 2-way analysis of variance with Dunnett’s post hoc test relative to WT.
Discussion
The discovery that loss of NBEAL2 expression causes GPS21-23 intensified efforts to determine the role of NBEAL2 in α-granule biogenesis, employing a variety of approaches.18,27,28,32,33,35,42,49 Several potential interactions of NBEAL2 with other proteins have been reported,41 but their roles in α-granule development remain largely unclear. We observed that MKs lacking NBEAL2 showed the loss of protein cargo from P-selectin-positive vesicles via RAB11-associated endosomal trafficking,35 indicating that NBEAL2 is required for nascent/mature α-granules to obtain and/or retain their cargo. Along with other observations,33 these findings indicate that the myelofibrosis often observed in GPS patients can be attributed to the loss/secretion of growth factors and other α-granule cargo from NBEAL2-null MKs into the extracellular bone marrow. These factors can attract fibroblasts and other cells, triggering fibrosis that gradually replaces hematopoietic cells.
Recent studies have shown that the ER extends throughout cells, providing extensive opportunities for interactions that facilitate intracellular signaling, ion homeostasis, lipid transfer, organelle remodeling, and trafficking.36-39 Many of these processes have been linked to α-granule development, making it highly likely that the extensive ER of MKs plays a key role in granule biogenesis. ER-resident proteins that have been linked to this process include SEC22B, reported to interact with VPS33B.31,41 The 24.5-kDa SEC22B protein contains a C-terminal transmembrane domain from which SNARE coiled-coil and N-terminal longin domains50 extend into the cytoplasm. It is postulated that the SEC22B longin domain remains open for SNARE complex assembly and is able to fold back onto the coiled-coil domain for incorporation into coat complex II–coated vesicles that transport protein from the ER to Golgi.50 SEC22B has been reported to have both vesicular and nonvesicular functions in organelle maturation and membrane expansion43,44,51 and to function as a vesicular-SNARE (or R-SNARE) during both anterograde and retrograde transport of vesicles between the ER and Golgi.43,52-55 Additional roles include endosomal recycling–mediated antigen cross-presentation56 and phagosome maturation in dendritic cells, where SEC22B depletion inhibits the recruitment of ER-resident proteins to phagosomes and vacuoles containing Toxoplasma gondii parasites.51 SEC22B is also required for plasma membrane expansion during neuronal development,44 autophagosome biogenesis,57 secretory autophagy,58 very low-density lipoprotein secretion,59 and early embryonic murine development.60
SEC22B has been shown to interact with VPS33B,31,41 but the precise intracellular localization of this interaction has not been explored. SEC22B has been localized to the ER-Golgi intermediate cellular compartment51,56 ; however, SEC22B is not restricted to this ER compartment.44 In an initial immunofluorescence microscopy–based screen, we observed extensive overlap of SEC22B with NBEAL2 in mature human MKs (Figure 1E). Direct interaction of these proteins was confirmed via co-IP experiments using tagged proteins expressed in HEK293 cells (Figure 1A-B). This led us to investigate whether this interaction can occur in human MKs, and we confirmed that endogenous NBEAL2 and SEC22B coimmunoprecipitate from MK lysates (Figure 1C-D). Because SEC22B is an abundant ER-resident protein with multiple functions, it is likely that only a subset of molecules interact with NBEAL2. This expectation is consistent with our immunofluorescence microscopy imaging of human MKs, which indicated overlap of SEC22B with NBEAL2 outside of a central region we suspected to be the protein synthetic ER/Golgi (Figure 1E). To more precisely delineate where NBEAL2 and SEC22B interact within human MKs, we examined several ER-resident proteins (Figure 2). Consistent with our earlier study, we observed strong overlap of NBEAL2 with P-selectin and weak overlap of NBEAL2 with calnexin, a chaperone largely restricted to the protein-synthesizing ER.35 NBEAL2 showed much stronger overlap with the calcium channel SERCA3, abundant in the outer ER,61 and with SEC22B. Therefore, our data indicate that NBEAL2 interacts with SEC22B in the outer ER of human MKs, where SEC22B is involved in vesicular trafficking and membrane contact site formation.
NBEAL2 is a member of the BEACH domain–containing protein family, with 9 known members.26 One of these is lysosomal trafficking regulator (LYST), where mutations result in abnormal intracellular lysosomal transport, causing the giant cytoplasmic granules in neutrophils and eosinophils that are the hallmarks of Chediak-Higashi syndrome (OMIM: 606897).62,63 These observations indicate a role for LYST in vesicular trafficking, but additional details have remained elusive in the 2 decades since. NBEAL2 is also a large BEACH protein with multiple putative functional domains28 (Figure 3A). How these or other parts of the protein may be involved in cellular trafficking events is also largely unknown.
We clarified the binding interaction of NBEAL2 with SEC22B by coexpressing tagged constructs containing full-length SEC22B and various sections of NBEAL2. We first examined the N-terminal portion containing the armadillo-like and concanavalin A–like domains (N1) and a C-terminal portion containing the PH-BEACH and WD40 domains (N2) and observed binding of only the latter to SEC22B (Figure 3A-B). Further delineation of the binding site revealed that SEC22B binds to a 106 aa region encompassing residues 1798 to 1903 of NBEAL2 (N5; Figure 3A-B). Although this region does not encompass a specific functional domain, it does contain the locations of 2 missense mutations previously found in patients with GPS (Figure 4A). This suggested that the NBEAL2 missense mutations may abrogate binding with SEC22B. This was confirmed via co-IP experiments where NBEAL2 constructs containing either E1833K or R1839C failed to bind SEC22B (Figure 4B). Both variants were able to bind P-selectin, as was WT NBEAL2, as we previously demonstrated35 (Figure 4C). Thus binding of SEC22B to NBEAL2 via the aa 1798 to 1903 region may be important for α-granule biosynthesis.
NBEAL2 is predicted to be a cytoplasmic adaptor that may bridge membrane-associated proteins, such as SEC22B on the ER and P-selectin on α-granules. We examined the possibility of a tripartite interaction by coexpressing SEC22B and P-selectin in the presence or absence of NBEAL2. We observed that SEC22B only coimmunoprecipitated P-selectin in the presence of NBEAL2 in HEK293 cells (supplemental Figure 2) or in imMKCL cells (Figure 4D), indicating that NBEAL2 can bind both partners simultaneously. This suggests that NBEAL2 may function as a tether that links α-granules to the ER via interactions with SEC22B and P-selectin.
To determine whether SEC22B is important for MK α-granule formation, we made use of imMKCL cells established by the Eto group.46 These cells have been used for ex vivo platelet production,48,64 and previous studies have confirmed they produce α-granules as visualized by transmission electron microscopy (eg, Figure S1D in the report by Ito et al48 ; Figure 5A; supplemental Figure 4 WT). Using CRISPR/Cas9 gene editing, we were able to knock out NBEAL2 and SEC22B expression in imMKCL cells, with SEC22B-KO and NBEAL2-KO cells showing complete loss of their respective proteins (Figure 6B). Both KOs showed total loss of α-granules, which were abundant in both WT and mock-transduced cells (Figure 5C-D; supplemental Figures 4 and 5A). Therefore, like NBEAL2, SEC22B is required for α-granule production.
KO of either NBEAL2 or SEC22B did not affect mitochondria numbers in imMKCL cells (Figure 5C-D; supplemental Figures 4 and 5B). Transmission electron microscopy suggested that the demarcation membrane system may be altered with SEC22B and NBEAL2 loss (Figure 5; supplemental Figure 4), as also observed in NBEAL2-KO mice.27 δ-granules could not be accurately evaluated in imMKCL cells, where whole-mount electron microscopy of platelets would be more informative.65,66 It was possible to examine the effects of loss of SEC22B on ER structure via immunofluorescence microscopy. The distributions of calnexin and SERCA3 indicated the overall ER architecture was not overtly altered in either SEC22B-KO or NBEAL2-KO cells (supplemental Figure 3). Both calnexin and SERCA3 were observed to colocalize with SEC22B in WT and NBEAL2-KO imMKCL cells (supplemental Figure 3A-B). Therefore, loss of SEC22B expression did not cause major changes in ER configuration in imMKCL cells.
Interestingly, the loss of SEC22B was accompanied by significant reduction in NBEAL2 expression, but not vice versa (Figure 6B). This suggests that the expression and/or stability of NBEAL2 is dependent on the presence of SEC22B. The observation that SEC22B expression levels are not affected by loss of NBEAL2 can likely be attributed to the many functions of SEC22B that do not involve NBEAL2. In addition to the loss of α-granules, we also noted substantial decreases in the amounts of P-selectin and the α-granule cargo proteins von Willebrand factor and thrombospondin-1 present in lysates from SEC22B-KO and NBEAL2-KO imMKCL cells (Figure 6B-C). Expression of these proteins returned to WT levels in KO cells that were rescued by introducing expression of tagged versions of NBEAL2 or SEC22B (ie, NBEAL2-R and SEC22B-R cells; Figure 6B-C). These observations are in keeping with the previously observed loss of granule cargo proteins from NBEAL2-KO mouse MKs.27,35
How do these findings relate to the production of α-granules by MKs? As indicated earlier, we believe that VPS33B/VPS16B are involved early in α-granule development, likely acting at a trans-Golgi compartment to facilitate formation of granule precursors, because loss of either VPS33B or VPS16B completely blocks α-granule production.24,25 NBEAL2 can bind P-selectin,35 and it is required to prevent MK vesicles containing this protein from losing their cargo via RAB11-mediated secretion.35 The ER plays a key role in endosome maturation,36-39 and the side of the ER facing α-granules has abundant SEC22B that can function as a fusogenic or nonfusogenic SNARE.50 Its nonfusogenic function may stabilize close contacts between the ER and plasma membrane to facilitate nonvesicular lipid transfer.44 We propose that NBEAL2 interacts with P-selectin on precursor/mature α-granules and with SEC22B on the ER to facilitate interactions essential for α-granule development and stability. Loss of SEC22B may disrupt this interaction, leading to failure of α-granule development and degradation of NBEAL2. A model of α-granule biogenesis incorporating these proposed features is shown in Figure 7. ER extending throughout MKs interacts with Golgi, trafficking vesicles, α-granules, and many other structures not shown (eg, mitochondria, peroxisomes, endosomes and the plasma membrane). SEC22B facilitates interaction of α-granules with the ER via binding to NBEAL2, which also binds P-selectin present on nascent/mature granules. The functional roles of this tripartite interaction remain to be determined, and nonexclusive possibilities range from granule tethering to lipid transfer via membrane contact sites.
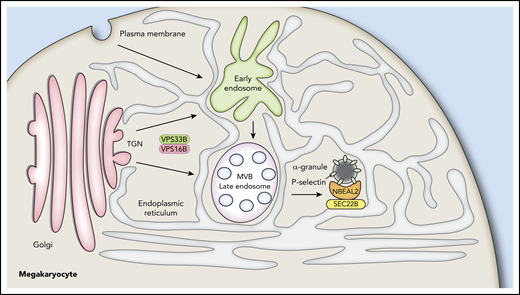
Proposed interactions of NBEAL2 with SEC22B and P-selectin during α-granule biogenesis. The ER extends throughout MKs, making numerous contacts with the Golgi, organelles, early endosomes, late endosomes/multivesicular bodies (MVB), and other vesicles. α-granule biogenesis likely begins with events at the trans-Golgi network mediated by the VPS33B/VPS16B complex and proceeds via maturation of endosomes into α-granules containing the membrane spanning protein P-selectin. We showed previously that NBEAL2 can bind to P-selectin and that these proteins likely interact on precursor and/or mature α-granules to stabilize their protein cargo.35 We show in this study that NBEAL2 can simultaneously bind to SEC22B and present evidence this interaction may be required for α-granule formation. A possible implication of these interactions is that NBEAL2 acts as a bridge between the ER and forming/mature α-granules that is necessary for granule production and/or cargo retention.
Proposed interactions of NBEAL2 with SEC22B and P-selectin during α-granule biogenesis. The ER extends throughout MKs, making numerous contacts with the Golgi, organelles, early endosomes, late endosomes/multivesicular bodies (MVB), and other vesicles. α-granule biogenesis likely begins with events at the trans-Golgi network mediated by the VPS33B/VPS16B complex and proceeds via maturation of endosomes into α-granules containing the membrane spanning protein P-selectin. We showed previously that NBEAL2 can bind to P-selectin and that these proteins likely interact on precursor and/or mature α-granules to stabilize their protein cargo.35 We show in this study that NBEAL2 can simultaneously bind to SEC22B and present evidence this interaction may be required for α-granule formation. A possible implication of these interactions is that NBEAL2 acts as a bridge between the ER and forming/mature α-granules that is necessary for granule production and/or cargo retention.
To conclude, our observations show that the ER-resident protein SEC22B is required for α-granule biogenesis, and it can bind NBEAL2 via a specific region (aa 1798-1903) where the presence of either of 2 GPS-associated missense variants (E1833K and R1839C)22 abrogates the interaction. We also observed that NBEAL2 has the potential to simultaneously bind both SEC22B and the α-granule membrane protein P-selectin. This tripartite interaction may facilitate processes involved with α-granule maturation, where the precise functional roles of SEC22B and the ER remain to be determined.
Contact the corresponding author for original data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Kirin Brewery Company for its generous gift of recombinant pegylated human MK growth and development factor (thrombopoietin).
R.W.L. is supported by the RESTRACOMP fellowship from the Research Institute of the Hospital for Sick Children, Toronto, Ontario, Canada. W.H.A.K. is supported by operating grants from the Canadian Institutes of Health Research (MOP-119450 and PJT153168).
Authorship
Contribution: R.W.L., L.L., R.L., and F.G.P. carried out investigations under the supervision of W.H.A.K.; K.E. provided the imMKCL cells; R.W.L. and W.H.A.K. wrote the manuscript with contributions from F.G.P.; and all authors agreed to be included as coauthors of this manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Walter H. A. Kahr, Departments of Paediatrics & Biochemistry, University of Toronto, Division of Haematology/Oncology, Cell Biology Program, The Hospital for Sick Children, 555 University Ave, Toronto, ON, M5G 1X8, Canada; e-mail: walter.kahr@sickkids.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal