


Abstract
Natural killer cell deficiencies (NKDs) are an emerging phenotypic subtype of primary immune deficiency. NK cells provide a defense against virally infected cells using a variety of cytotoxic mechanisms, and patients who have defective NK cell development or function can present with atypical, recurrent, or severe herpesviral infections. The current pipeline for investigating NKDs involves the acquisition and clinical assessment of patients with a suspected NKD followed by subsequent in silico, in vitro, and in vivo laboratory research. Evaluation involves initially quantifying NK cells and measuring NK cell cytotoxicity and expression of certain NK cell receptors involved in NK cell development and function. Subsequent studies using genomic methods to identify the potential causative variant are conducted along with variant impact testing to make genotype-phenotype connections. Identification of novel genes contributing to the NKD phenotype can also be facilitated by applying the expanding knowledge of NK cell biology. In this review, we discuss how NKDs that affect NK cell cytotoxicity can be approached in the clinic and laboratory for the discovery of novel gene variants.
Introduction
Natural killer (NK) cells are innate immune lymphocytes critical in the defense against virally infected cells and cancer. Their function includes contact-dependent targeted cell lysis through the specific delivery of highly specialized organelles termed lytic granules, containing pore-forming perforins and apoptosis-inducing granzymes, or through death receptor–based signaling.1 Unlike CD8+ T cells, which use a T-cell receptor for specific target recognition, NK cells rely on a balance of activating and inhibitory signals through germ line–encoded cellular receptors, giving them a unique role in the early stages of immune response. In both murine models and human contexts, NK cells have demonstrated value in the defense against herpesviruses and in surveillance of cancer cells.2-4
As with other immune cells, deficiencies in NK cell development and function have been identified and lead to disease susceptibility. NK cell deficiencies (NKDs) are considered a phenotypic subtype of primary immunodeficiency diseases (PIDDs), where the major immunological issue resulting in a clinical immunodeficiency is an NK cell abnormality.5-7 Diagnosis of NKDs begins with a clinical suspicion for an NK cell defect, where patients often present with recurrent, atypical, and/or severe infection by herpesviruses, such as Epstein-Barr virus, cytomegalovirus, varicella zoster virus, and herpes simplex virus, as well as human papillomavirus. This clinical observation is a critical theme in the consideration and diagnosis of the NKD phenotype and should prompt further diagnostic attention. Other PIDDs can include a defect of some kind in NK cells and can also include herpesviral susceptibility (as discussed elsewhere),5,7-9 but it is only when the immune defect and clinical immunodeficiency resulting from NK cell abnormality represent the primary immunological impairment that they are consistent with the NKD phenotype. An example of a PIDD with an NK cell abnormality is severe combined immunodeficiency resulting from an aberration of the IL2RG gene, where NK cells as well as T cells fail to develop. Here the latter represents the major immunological defect resulting in clinical immunodeficiency. This is notably different from NKDs, which are associated with a specific genetic differential diagnosis and have the potential to uncover genes and variants more specifically affecting NK cells, thereby enriching our understanding of NK cell biology.
NKDs can be considered in 2 categories: classical and functional (Figure 1). In classical or developmental NKD, the development, production, or survival of NK cells is perturbed, leading to a reduced cell number, whereas in functional NKD, NK cell number and maturity are normal but some aspect of their function is defective. Currently, 5 genes have been at least in part attributed to the differential for classical NKD: GATA2, MCM4, RTEL1, GINS1, and IRF8.7,10-15 All classical NKDs include deficits in NK cell function; however, these deficits result from impaired development or maturation of circulating NK cells. Many patients with defects in these genes have other immunological abnormalities and as such do not have an NKD, as is the case with GATA2 deficiency, where many patients present with B-cell, dendritic cell, and CD4+ cell abnormalities, suggesting the presence of other complex genetic and environmental interactions unique to each patient. However, among the patients studied, there is at least a subset who have presented with the NKD phenotype, including focused clinical presentations of recurrent/severe herpesviral infection. This remains a common theme for these patients, again with at least a subset of patients affected by each gene having the NK cell abnormality as the primary immunological deficiency and phenotype.7,16 Therefore, these genes are considered part of the differential for NKDs, although identification of an aberration in 1 of these genes in no way implies the NKD phenotype.
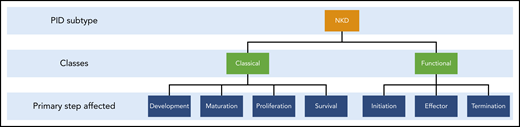
The 2 classes of NKDs and the critical steps of development and cytotoxic function. The focus of this review is on the functional arm.
The 2 classes of NKDs and the critical steps of development and cytotoxic function. The focus of this review is on the functional arm.
Although the identification of functional NKD has been less frequent than that of classical NKD, it is probable that there will be additional causes discovered through genetic and biological analyses of NKD patients. In this review, we will focus on functional aberrations of NK cells and how investigations into NK cell cytotoxic biology can guide our study of possible novel genes that are causative of NKDs as well as facilitate novel gene discovery.
Human NK cell cytotoxicity
NK cells perform cytolysis through a series of linear cell biological steps. After undergoing licensing through the recognition of self–major histocompatibility complex class 1 molecules, NK cells are permitted to perform directed cytolysis of target cells.17,18 This occurs in 3 phases: initiation, effector, and termination, whereby the NK cell engages with its target, induces cell death in the target, and detaches to proceed to the next target.19
In the initiation phase, NK cells combine cell migration with sensing and subsequently engage in target cell adhesion. This stage involves recognition through the balance of activating and inhibitory signals by receptors including DNAM-1, NKG2D, natural cytotoxicity receptors (eg, NKp46, NKp44, and NKp30), KIR, and LFA-1, all shown to be important in cellular cytotoxicity.20-25 This phase also involves movement of lytic granules toward the microtubule organizing center via dynein motor protein and associated adaptors.23,26-28
In the effector phase, the NK cell proceeds to kill the target cell by forming a mature immune synapse (IS), whereby actin accumulates at the interface between the NK cell and target.29,30 The actin reorganization is tightly regulated and composed of an initial formin-based polymerization followed by an Arp2/3-mediated polymerization.31 Proteins involved in these coordinated events include GTPases such as CDC42, Ras, and Rac, motor proteins such as myosin IIA, and associated adaptors and regulators such as IQGAP1, WASP, DOCK8, EVL, and CIP4 in NK cells as well as RAB27A, LYST, MUNC13-4, and the Syntaxin family for lytic granule formation and movement.32-42 In addition, outside-in signaling by LFA-1 and other surface receptors modulate actin and microtubule dynamics and microtubule organizing center (MTOC) positioning.23,31,43 Firm adhesion to the target is followed by lytic granule delivery by MTOC polarization toward the IS.44,45 Although aberrations of these are likely to affect both NK cells and cytotoxic T lymphocytes (CTLs), there are theoretically some proteins that are specific to NK cells and variants that will have predominant impacts upon them.
After delivery to the IS, lytic granules must traverse a contiguous yet dynamic actin mesh before granule docking and exocytosis. Granules proceed, presumably stochastically, through size-permissible clearances modulated by Arp2/3-mediated actin branches for subsequent membrane fusion and lytic cargo release into the synaptic cleft. Additionally, myosin 2A regulates lateral motion of synapse actin fibers.31 In Chediak-Higashi syndrome, where LYST mutation leads to enlarged lytic granules and accompanying reduction of lytic granule degranulation, the actin mesh similarly inhibits granule passage, indicating an active regulatory process.41
Various motors are critical for the effector stage, including the dynein motor, the myosin 2A motor, and various kinesin motors.28,46,47 They modulate lytic granule mobility toward and away from the MTOC and possibly on and through the actin mesh at the IS. In addition, various motor regulators/adaptors have been identified, such as HkRP3 and VASP.48,49 Therefore, it is reasonable to assume that defects in associated regulatory genes can lead to an NKD only if these genes are preferentially expressed in and used by NK cells and/or the mutation affects an NK cell–specific function, as is the case with FCGR3A (discussed in “Elucidating mechanisms of disease in patients with NKDs”).
In the termination phase, detachment from the target cell and subsequent engagement with another target occurs, termed serial killing. Detachment and efficient serial killing are dynamic processes dependent on intact CD16 functioning and subsequent shedding and membrane receptor recycling.50,51 Serial killing also serves as a unique transition point from the initial kill, as the NK cell switches from a granzyme B–based to a death receptor–mediated cytotoxicity.1,52 Together, these 3 stages offer critical regulatory nodes that would be susceptible to NK cell–specific mutations that can lead to potential functional NKDs.
Clinical approach to the diagnosis of NKD
The identification and understanding of novel causes of NKDs are dependent on careful clinical and biological study. Upon suspicion of an NKD (and in concert with exclusion of other known broader immunodeficiencies), patient peripheral blood is typically assessed for cytotoxic function and NK cell phenotype. Patient peripheral blood mononuclear cells (PBMCs) are typically cocultured with major histocompatibility complex class 1–deficient target cells for the measurement of natural cytotoxicity or target cells susceptible to antibody-mediated cellular cytotoxicity after opsonization with a monoclonal antibody. From this, a determination can be made as to whether a cytotoxic deficit lies in the ability to kill susceptible targets, those that require the CD16 (FcγRIIIa) receptor for recognition, or both. Should a functional defect be identified, the stability of this impairment should be established by repeated assays at spaced intervals. Because testing in most clinical diagnostic contexts is performed using PBMCs, the relative percentage of NK cells within the mixture of mononuclear cells needs to be considered. A very low percentage of NK cells can artificially make function appear deficient, whereas individual NK cell function might be intact. In these cases, purity enrichment of NK cells followed by cytotoxicity testing can be definitive. Other functional assays can also be useful and agnostic to the percentage of NK cells among PBMCs, such as CD107a upregulation detected using flow cytometry on NK cells directly.53 Although not a measure of cytotoxicity, CD107a upregulation measures degranulation after activation, which in most cases correlates with cytotoxic function. It is critical to appreciate, however, that CD107a upregulation can be normal, but cytotoxicity can still be defective, such as in the case of perforin deficiency (which is another example of a PIDD that includes an NK cell abnormality but is not an NKD). Other NK cell defects yet to be discovered will also likely emphasize this dichotomy, such as those that would prevent directionality in degranulation or have other aberrations of lytic granule contents.
In addition to functional testing, cell phenotyping by flow cytometry allows for assessment of NK cell frequency and detailed subset distribution. The measurement of the number of CD56+CD3− NK cells in peripheral blood enables the calculation of lytic units estimating NK cell killing capability. More exhaustive phenotyping includes the assessment of markers of NK cell development and maturation by evaluating activation-, inhibitory-, and development-associated receptors.7,54 Although not the focus of this overview, classical NKD that affects development can be distinguished using detailed developmental phenotyping as derived from the immunological studies of human NK cell development.7 If either NK cell number and/or function is impaired, further evaluation is suggested and described in “NKD vs HLH” and "Elucidating mechanisms of disease in patients with NKDs." Management generally involves treatment of the viral infection and subsequent lifelong antiviral prophylaxis. The formal, detailed diagnostic approach and treatment of NKDs and larger topic of PIDD management and discovery are discussed in other overviews.6,55,56
NKD vs HLH
Although both can be associated with impaired NK cell cytotoxicity, NKDs are distinct from hemophagocytic lymphohistiocytosis (HLH).57 HLH can be confused with an NKD, because HLH is diagnosed based on the HLH 2004 criteria, which include the presence of defective NK cell cytotoxicity, and is usually classified as either primary (familial) or secondary (with respect to some other underlying etiology).58 Although only some HLHs include a defect of NK cell cytotoxicity, even when cytotoxicity is impaired it also includes defects in CTLs, and the mechanism resulting in clinical disease is distinct from that in NKDs, leading to a very different clinical history and presenting phenotype. More recently, genetically explicable HLH has been described as being associated with either a cytotoxicity-based inability to dampen inflammation or an inherent cell-intrinsic inability to restrain inflammation (eg, an inflammasome abnormality).57,59 In either case, HLH is phenotypically driven by a toxic activation of the immune system resulting from an inability to either eradicate or dampen response to a stimulus, such as a virus. In the varieties with lytic abnormalities, uncontrollable systemic inflammation occurs, because cytotoxic cells are unable to eradicate the source of the antigenic stimulus (eg, a virus-infected cell) to curtail an otherwise appropriate immune response. For example, patients with perforin deficiency have an activated immune response, leading to profound inflammation and immune dysregulation. However, the patient’s disease results from an inflammatory response that cannot be controlled because of the loss of T and NK cytolytic function and is exacerbated by the patient’s own intact and excessive cytokine production and stimulation of dendritic cell responses.60-63 Therefore, the issue here is not viral replication per se, but rather defective viral control and excessive stimulation and inflammation.
In contrast, NKD patients typically present with recurrent, severe, or atypical consequences of infections by herpesviruses (consequences of poorly controlled viral replication) and the majority the immunological defect is in NK cell compartment, leading to the clinical immunodeficiency.5 However, confusion lies in comparing NKD to cytotoxic HLH, because both can present with a defect in NK cell function using standard cytotoxicity assays. Again, in cytotoxic HLH, both NK cells and CTLs are defective in cytotoxicity but not cytokine activity, leading to an inability to eradicate infected cells that are promoting immune responses and inflammation. With NKDs, the NK cell abnormality serves as the primary immunological defect leading to the clinical presentation, and CTL function is usually preserved. Abnormalities in other immune compartments can be present, but these are present in a minority of cases and are not the main drivers of the observed clinical phenotype. They do, however, still provide important biological insights into gene function in different cellular contexts. NK cytotoxicity assays, CD107a upregulation, CTL function, NK cell phenotyping, and genomic sequencing are critical to parsing out the underlying biology and separating the 2 entities and have been previously described.53,57 NKDs, while impairing cytotoxicity, are distinct from HLH and in general will not cause HLH, because CTL function will remain intact and compensate to prevent the toxic inflammation characteristic of HLH. In this light, NKDs illustrate the specific utility of NK cells in human host defense and underscore their particular role distinct from CTL defenses, particularly in the context of herpesviral infections.
Elucidating mechanisms of disease in patients with NKDs
The current paradigm in identifying novel NKD genes is primarily patient driven and has been mostly, and at least initially, based on single patients having suggestive clinical and biological phenotyes. However, a variant in a gene known to be relevant to NK cell function in a patient with a suggestive clinical phenotype is not sufficient to define causality. This must be accomplished using a process for biologically and genetically establishing mutation impact and variant causality through satisfying key criteria.64 To attribute an observed clinical phenotype to a particular genotype, studies must show the genotype is monogenic and does not occur in individuals without the observed clinical sequelae. Furthermore, the variant must affect the gene product in either expression or function, and causality should be shown in a relevant in vitro or in vivo model. Studies must demonstrate the variant is unique and rare and can be recapitulated in a patient-relevant system. Performing genomic sequencing and associating observed symptoms with a rare or previously undescribed variant are again insufficient to declare mutation impact or causality, so additional mechanistic studies are always required. Exome and genome sequencing in combination with in silico tools for variant impact predictions serve as an initial platform for guiding in-depth analysis but cannot be confidently linked to the clinical phenotype without biological studies. Therefore, these criteria have been the foundation of the discovery of many novel gene variants causative of PIDDs and the underlying mechanisms of known PIDDs for treatment development.41,65
After screening (cytotoxicity and phenotype) analyses, if a patient is deemed a candidate for a possible NKD, unbiased genomic sequencing and in-depth clinical and biological analyses, including pedigree analysis and mechanistic studies, should be performed to establish a molecular diagnosis. Exome sequencing has allowed for the identification of novel PIDD genes, especially in cases where familial linkage occurs.66,67 After exome sequencing, in silico tools (eg, eXAC browser, CADD, MCAP, SIFT, Polyphen2, and MutationTaster)68-70 are initially used to assess variant prevalence and the impact/deleteriousness of both the amino acid and nucleotide changes on gene stability, and gene product structure and function are used to identify possible gene candidates in an unbiased filtering approach.66 Then, additional analyses are performed where each gene is further evaluated in the context of existing literature and databases for connections to the observed patient characteristics or the mechanism that could be underlying the NKD phenotype. Subsequently, patient modeling and functional studies are performed, guided by known or predicted roles of candidate genes, to establish a biological basis, confirm a molecular diagnosis, and in the best case develop a possible therapeutic strategy.
The FCGR3A (CD16) homozygous p.L66H variant leads to defective NK cell cytotoxic function and is an example of how perturbations that affect a pathway or interaction specific to NK cell cytotoxicity can lead to functional NKDs. Three unrelated patients who presented with recurrent infections by herpesviruses, including herpes simplex virus and Epstein-Barr virus, demonstrated normal CD16 expression but reduced binding of the B73.1 monoclonal antibody to CD16, which was suggestive of an NK cell–specific defect in the B73.1 epitope region.71-73 Additionally, patients had a defect in NK spontaneous but not antibody-dependent cellular cytotoxicity (ADCC).71 CD16 has been primarily associated with ADCC owing to its ability to bind IgG, but the L66H variant alters the structure of the first immunoglobulin G–like domain of CD16 that was determined through studies of the patient-derived variant to be important for interaction with CD2, an NK cell costimulatory receptor.71 As a result, in patients with homozygous CD16 variants, CD2 engagement is not able to leverage CD16 signaling for spontaneous killing while CD16 expression and function remain intact for ADCC.
To date, the homozygous p.L66H variant in FCGR3A is the sole known cause of functional NKD.71 One hypothesis for the imbalance of classical vs functional NKD genes is that NK and T cells share many genes involved in cytotoxic function. Therefore, to cause functional NKD, a mutation must specifically perturb an NK cell–specific pathway. Theoretically, this could occur through a protein that is nonredundant in NK cells yet dispensable in other immune cells or a protein used only by NK cells for specifically cytotoxic function. An aberration leading to functional NKD could even conceivably occur in a microRNA or a protein extrinsic to NK cells, although these have not yet been identified. Furthermore, functional NKD is more challenging to clinically detect, because the main clinical diagnostic study for evaluating NK cells is their enumeration via flow cytometry. Of course, the exploration of patients with an NKD phenotype may result in the discovery of a defect belonging in the broader category of PIDD with an NK cell abnormality, especially as more patients with a similar defect are identified or if the clinical evolution of a patient’s phenotype suggests a broader defect.
We do, however, expect the emergence of more functional NKDs as the understanding of NK cell cytotoxic biology expands and more phenotypically relevant patients are actively considered. We are aware of ongoing work regarding genes involved in NK cell function and potentially causative of NKDs, such as ATP6V0A2.74 Also, given the rarity of functional NKDs, general growth in NK cell biology will facilitate both proactive and reactive targeted studies to accelerate genetic and molecular diagnoses by establishing critical investigative points.
Research approach to identifying novel genes important in NK cell biology and variants causative of NKDs
To study both the mutational impact and the molecular underpinnings of an NKD phenotype, studies can be split into 5 components: (1) genetic analysis, (2) phenotypic analysis, (3) biochemical analysis, (4) modeling, and (5) functional testing (Figure 2). Pairing this patient-guided research approach with basic NK cell biology guides laboratory studies of NK cell defects.
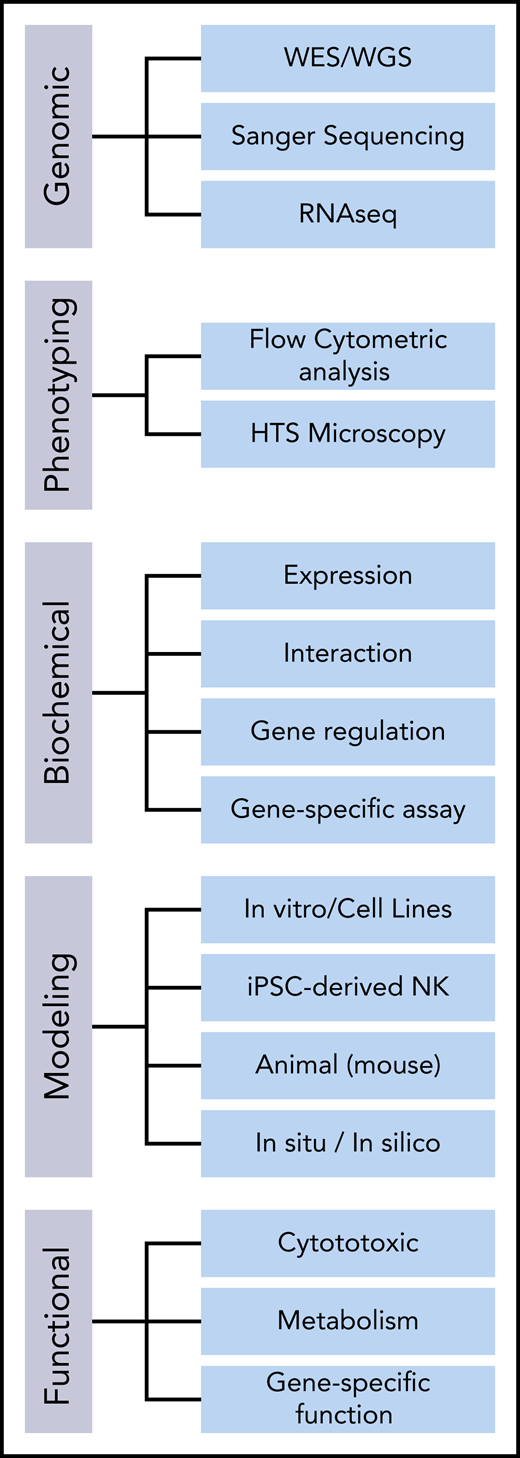
General approaches to studying the mutational impact and genotype-phenotype causality in candidate genes for NKDs. HTS, high-throughput screening; iPSC, induced pluripotent stem cell; RNAseq, RNA sequencing; WES, whole exome sequencing; WGS, whole genome sequencing.
General approaches to studying the mutational impact and genotype-phenotype causality in candidate genes for NKDs. HTS, high-throughput screening; iPSC, induced pluripotent stem cell; RNAseq, RNA sequencing; WES, whole exome sequencing; WGS, whole genome sequencing.
Genetic and predictive analysis
As exome, genome, and transcriptome sequencing continue to become more affordable and accessible, targeted genetic analysis is often performed as a first step and can replace or be complementary to an unbiased approach for rapid diagnosis and an accelerated research pipeline.14,57,67,75 After establishing a candidate gene variant, the variant should be confirmed by Sanger sequencing, and biallelic variants should be validated as occurring in trans (and not on the same allele).14 Identification of variants can then be complemented by modeling of the protein structure to determine the predicted effect on function. Additional resources include existing databases of tissue-specific gene and protein expression. Although the relative expression of a gene of interest in primary NK cells should not ultimately determine its likelihood as a disease-causing variant, it can affirm that previously undefined proteins may play a role in NK cell function.
Phenotypic analysis
Adhesion, activation, and inhibitory receptors are critical to NK cell function and offer important theoretical disease-associated variants. Flow cytometric analysis can assess expression defects in activating and inhibitory receptors and serve as an initial assessment tool for surveying the overall receptor environment (when compared with an adequate range of age-matched controls). In addition, as more advanced quantitative microscopic methods for assessing the localization and distribution of these receptors as well as immune synapse architecture emerge, they can serve as a critical evaluative method that, along with newer technologies, is even feasible for screening.31,35,76,77
Biochemical analysis
Initial biochemical testing of candidate gene variants involves assessing protein expression. Reduced expression levels can be indicative of a deleterious mutation leading to nonsense mediated decay or protein instability.78 Elevated protein expression levels can be indicative of a variant protein acquiring a novel function or altered gene regulation that is ultimately detrimental to cellular function. If relative protein levels are comparable with those of a healthy donor, this can still indicate a pathogenic process if the protein is indeed translated but is dysfunctional. In this case, studies assessing the relevant physiological interactions of the protein should be performed to search for defects in protein-protein interactions and downstream functional effects, as nicely demonstrated for some of the genes causing HLH.32,40 This can include conventional biochemical approaches such as coimmunoprecipitation, reporter assays, and phosphorylation-specific protein analyses. In conjunction, cell- or animal-based modeling and accompanying functional testing should be performed.
Cell-based and in vivo modeling
To make genotypic and phenotypic correlations, model systems carrying the candidate gene variant should be used to recapitulate the observed patient phenotype. Models range in complexity from cell lines and iPSC-derived NK cells to animal modeling and human samples.79-81 Cell lines allow for high-capacity biochemical and functional studies in a relevant homogeneous clonal system. Although aberrant genes can be modeled in typical cell line systems like HEK293T cells,15 it is important to additionally consider using NK cell lines for modeling to provide an NK cell–related context as well as the ability to study effects on NK cell–specific functions. Established NK cell lines include NK-92, YTS, NKL, NK3.3, and KYHG-1, and these have been used to model NKDs through expression of the variant protein or endogenous modification using gene editing.71,82-84 These cell lines have distinct transcriptional and functional profiles85 ; therefore, selection of the right cell line to accurately recapitulate the observed defect is an important consideration. If patient samples are limited, producing patient-derived iPSCs followed by in vitro or murine in vivo differentiation to NK cells is a viable approach to model the patient-specific phenotype both during NK cell development and in subsequent functional assays of NK cell activities. This approach is in part enabled by the fact that human NK cells can be generated in vitro from hematopoietic precursors, a technique that has been used to model human NK cell development in the context of classical NKD.86,87 iPSCs have also been used in the context of PIDD, including to model and study hematopoietic development using patient-derived iPSCs from patients with GATA2 deficiency.88,89
For animal models, existing strategies include using gene introduction/alteration approaches to produce mice carrying the patient-relevant gene variant, which have been used to recapitulate the phenotype of other PIDDs. A standard approach is aimed at reproducing the mutation in mouse lines for subsequent assaying of mouse NK cells, which have been shown to be similar to human NK cells in certain aspects.14,90-95 More novel approaches use humanized mouse models to recreate particular human immune system compartments.96-98 To study NK cell functions, techniques such as adoptive transfer and the use of NK cell–specific promoters for compartment-specific expression can be employed.99 Other models, such as Caenorhabditis elegans and Danio rerio, are less complex but well characterized and can therefore be used to study gene regulatory pathways and organismal development as well as innate immunity more broadly and cell biology at an organism level.100,101 However, there is no true endogenous NK cell correlate in these models.
Functional assaying of NK cells
Functional evaluation of the biology underlying an NKD relies upon the extensive assessment of patient cells and the models created to further understand them. The mechanism by which a variant affects NK cell function can be systematically identified by dissecting the process of cytotoxicity itself and investigating each discrete step for impairment and subsequent downstream effects. Particularly by quantitative microscopy, each of the steps and stages of NK cell lytic function is a measurable, statistically assessable process. Although methods of analysis for some are well established, in other cases these may include deriving novel analytic approaches to quantify visualized proteins or cellular effects.
NK cell cytotoxic function should be assessed initially for intact conjugate formation with target cells, which is commonly performed by flow cytometric quantification. Formation of the IS is most frequently visualized by fixed-cell confocal microscopy, although other approaches, including recapitulation of lytic synapses on functionalized glass, can be informative. High-resolution microscopy, including live cell imaging, also enables the measurement of actin polymerization at the IS; lytic granule production, size, content, convergence, and mobility; and evolution of the synaptic interface (Figure 3).19,102-106 More specialized approaches, including electron microscopy and superresolution microscopy, have also been instrumental in defining the cellular phenotype of NK cells in HLH and PIDDs and will likely play a similar role in the definition of novel NKDs.41,103 In these approaches, it is important to distinguish CTLs from NK cells and keep in mind that to be considered an NKD, the disease must involve a patient with an immune phenotype or gene variant that is preferentially impactful to NK cells.
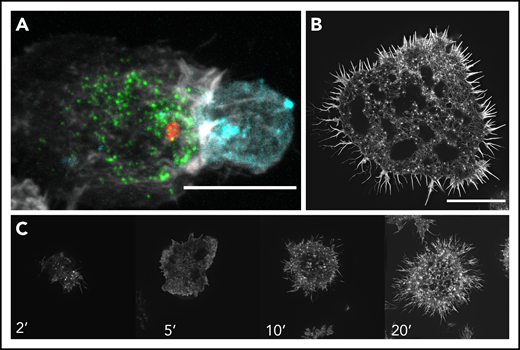
Examples of the NK cell IS during cytotoxicity. (A) Coincubation and subsequent fixed-cell imaging of NK cells (NK-92) conjugated to a K562 erythroleukemic target cell using spinning disk confocal microscopy. The image shows the formation of a lytic synapse between the NK and target cells where polymerized actin (white) is relatively enriched at the lytic immune synapse. The lytic granules (green; labeled using perforin as a marker for lytic granule content) are converged around the MTOC (red), with some having polarized toward the target cell (cyan; indicated with a membrane dye) as shown by the MTOC near the immune synapse. Image was cropped to focus on a single conjugate. (B) Structured illumination–total internal reflection fluorescence (SI-TIRF) microscopy of the actin meshwork at the IS of an NK cell (YTS) that was plated and fixed on an activating glass surface and then subsequently stained for F-actin using phalloidin. The activating surface is composed of anti-CD18 for adhesion and anti-CD28 for activation, thereby creating a representative target cell on a glass surface, enabling an en face assessment of actin architecture during IS formation. (C) Time lapse SI-TIRF microscopy of a YTS cell on an activated surface. YTS cells were placed on an activating glass surface and fixed at certain time points (2, 5, 10, and 20 minutes) and stained for F-actin using phalloidin to examine the evolution of the IS. The cell initially touches down upon the glass surface and initially extends out lamellipodia and filopodia as it spreads. By 20 minutes, a mature IS is formed as shown by the intricate dense actin mesh. Scale bars, 10 μm.
Examples of the NK cell IS during cytotoxicity. (A) Coincubation and subsequent fixed-cell imaging of NK cells (NK-92) conjugated to a K562 erythroleukemic target cell using spinning disk confocal microscopy. The image shows the formation of a lytic synapse between the NK and target cells where polymerized actin (white) is relatively enriched at the lytic immune synapse. The lytic granules (green; labeled using perforin as a marker for lytic granule content) are converged around the MTOC (red), with some having polarized toward the target cell (cyan; indicated with a membrane dye) as shown by the MTOC near the immune synapse. Image was cropped to focus on a single conjugate. (B) Structured illumination–total internal reflection fluorescence (SI-TIRF) microscopy of the actin meshwork at the IS of an NK cell (YTS) that was plated and fixed on an activating glass surface and then subsequently stained for F-actin using phalloidin. The activating surface is composed of anti-CD18 for adhesion and anti-CD28 for activation, thereby creating a representative target cell on a glass surface, enabling an en face assessment of actin architecture during IS formation. (C) Time lapse SI-TIRF microscopy of a YTS cell on an activated surface. YTS cells were placed on an activating glass surface and fixed at certain time points (2, 5, 10, and 20 minutes) and stained for F-actin using phalloidin to examine the evolution of the IS. The cell initially touches down upon the glass surface and initially extends out lamellipodia and filopodia as it spreads. By 20 minutes, a mature IS is formed as shown by the intricate dense actin mesh. Scale bars, 10 μm.
Future prospects in NKDs
With the advancing development of genomic technologies, microscopic techniques, and relevant cellular modeling that can recapitulate and probe a patient’s candidate gene variant, discovery and investigation of novel classical and functional NKDs will accelerate. By integrating research within the broader field of NK cell biology, it can be applied to NKD patients, allowing for targeted studies, especially where patient samples are limited. With the discovery of novel NKDs, it is hoped that advances in cellular therapeutics and methods to restore or enhance NK cell function in patients are on the horizon.
Authorship
Contribution: M.T.L., E.M.M., and J.S.O. wrote and reviewed the manuscript.
Conflict-of-interest disclosure: J.S.O. declares royalties from Wolters Kluwer Publishing (UpToDate) for chapters on NK cell deficiency. The remaining authors declare no competing financial interests.
Correspondence: Jordan S. Orange, Department of Pediatrics, Columbia University Medical Center, 622 W 168th St, New York, NY 10032; e-mail: jso2121@cumc.columbia.edu.
