

In this issue of Blood, demonstrate that driver mutations for human T-cell leukemia virus (HTLV)–associated adult T-cell leukemia lymphoma (ATLL) can be identified years before the clinical manifestations of disease are apparent.1
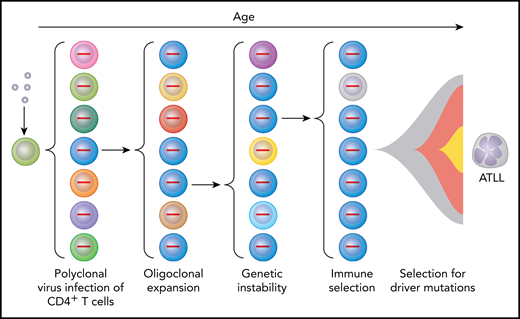
Multihit model for selection of HTLV-1–infected cells to become ATLL cells as a result of multiple intrinsic and extrinsic forces. Virus infection of CD4+ lymphocytes initially results in polyclonal infection (indicated by cells of different color with provirus shown in the nucleus). Expression of HTLV-1 Tax promotes oligoclonal expansion of infected T cells and genetic instability. Immune responses select for T cells with low levels of Tax expression. Driver mutations result in further selection resulting in ATLL in those individuals in which multiple “hits” have occurred to produce a lymphoproliferative malignancy.
Multihit model for selection of HTLV-1–infected cells to become ATLL cells as a result of multiple intrinsic and extrinsic forces. Virus infection of CD4+ lymphocytes initially results in polyclonal infection (indicated by cells of different color with provirus shown in the nucleus). Expression of HTLV-1 Tax promotes oligoclonal expansion of infected T cells and genetic instability. Immune responses select for T cells with low levels of Tax expression. Driver mutations result in further selection resulting in ATLL in those individuals in which multiple “hits” have occurred to produce a lymphoproliferative malignancy.
HTLV-1 has infected 5 to 20 million individuals.2 This retrovirus was identified 40 years ago as the cause of an aggressive T-cell malignancy.3 However, several key questions stymied researchers until recently. Why do only 2% to 5% of HTLV-1–infected individuals develop ATLL? Why does ATLL develop almost exclusively in individuals infected for many decades? How can one predict which individuals are at greatest risk of developing ATLL?
HTLV-1 is a retrovirus that encodes 2 oncoproteins, designated transactivator protein (Tax) and helix basic zipper protein (Hbz).4 Tax promotes the expression of viral structural and enzymatic proteins. Thus, viral particles are abundantly expressed from acutely infected cells, which spread to uninfected cells during the first several weeks after acquisition of infection (see figure). The viral envelope imparts tropism for CD4+ lymphocytes.5 Tax also promotes cell proliferation, resistance to apoptosis, and genetic instability. This results in the expansion of infected clones possessing various genetic defects. Hbz promotes expression of regulatory T-cell markers in infected cells.6 Thus, the number of infected CD4+ lymphocytes expands through clonal amplification over subsequent decades of infection, with selection of variants carrying mutations that promote T-lymphocyte proliferation and survival. Tax is also highly immunogenic, as are HTLV-1 structural and enzymatic proteins induced by Tax.7 Thus, selection also occurs for T cells producing very low levels of Tax.
A recent landmark description of the genomic landscape of ATLL provided answers to a number of questions in the field.8 This work showed a high rate of mutations in ATLL, consistent with the defects in DNA repair described in HTLV-1–infected cells.9 Particularly high rates of mutation were noted in the “Tax interactome” (ie, genes transcriptionally or posttranscriptionally activated by Tax), thus replacing the driving force of Tax.10 Mutations were identified in immune response genes, as expected from the antigenic properties of HTLV-1 proteins. Tumor suppressors were frequently inactivated, including TP53, CDKN2A, and Notch pathway genes. Particularly striking were gain-of-function mutations in genes involved in T-cell receptor and NF-κB pathway signaling, including phospholipase γ1, protein kinase Cβ, caspase-recruitment domain containing protein 11, and interferon regulatory factor 4. Moreover, there were frequent loss-of-function mutations or deletions in regulators of this pathway. Collectively, these genes were designated potential ATLL driver mutations.
The new study by Rowan et al has taken 1 huge step forward. Using blood samples from ATLL patients prior to presentation with disease, they identified potential ATLL driver mutations in all 6 ATLL patients, 2 to 10 years before disease onset. As a control, they used samples from 55 HTLV-1–infected individuals, with comparable provirus loads, who had not developed disease at a median of 10 years of follow-up and had a much lower frequency of driver mutations. This provides strong evidence that these mutations are specific for ATLL and could define a prognostic index. These findings provide an opportunity for making important advances for treatment of a disease that is usually fatal except for the few individuals who respond to allogeneic stem cell transplantation. These results also lead to numerous intriguing and important questions. Can one develop a sensitive and specific assay for predicting ATLL development based on the current findings, validated in a larger cohort of HTLV-1–infected subjects? Which mutations are most important? Is there a specific order for mutation selection? Can one prevent ATLL development by identification and treatment of individuals who have potential ATLL driver mutations? Is treatment directed at the T-cell receptor pathway effective for ATLL therapy or prevention? Can one use genomic landscape information to develop similar prognostic assays for individuals at risk of developing other leukemias and lymphomas? We look forward to answering these questions in the next few years.
Conflict-of-interest disclosure: L.R. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal