

In this issue of Blood, ask why a small proportion of red cells continues to produce fetal hemoglobin in adult life: importantly, they rule out some of the most favored previous explanations.1
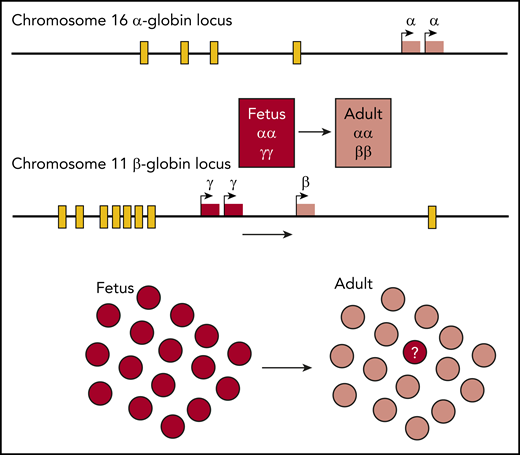
Hemoglobin is a tetramer comprising 2 α-like and 2 β-like globin chains encoded by genes in the unlinked α- and β-globin loci. Globin gene expression is regulated by distal enhancers (yellow boxes). The γ-globin gene is expressed during fetal life where it contributes to HbF but is silenced around the time of birth. However, normal adults express a small but variable amount of γ-globin in a small proportion of red cells called F cells. The question addressed here is what underlies γ-globin expression in the small numbers of HbF-containing cells. Answering this underlies strategies for therapeutic reexpression of γ-globin in adult life, which has the potential to ameliorate, or even cure, the major hemoglobinopathies, β-thalassemia and sickle cell disease.
Hemoglobin is a tetramer comprising 2 α-like and 2 β-like globin chains encoded by genes in the unlinked α- and β-globin loci. Globin gene expression is regulated by distal enhancers (yellow boxes). The γ-globin gene is expressed during fetal life where it contributes to HbF but is silenced around the time of birth. However, normal adults express a small but variable amount of γ-globin in a small proportion of red cells called F cells. The question addressed here is what underlies γ-globin expression in the small numbers of HbF-containing cells. Answering this underlies strategies for therapeutic reexpression of γ-globin in adult life, which has the potential to ameliorate, or even cure, the major hemoglobinopathies, β-thalassemia and sickle cell disease.
Over the past 50 years, erythropoiesis and globin gene expression have played a central role as a model to establish the principles by which mammalian genes are switched on and off during development and differentiation. In addition, investigating how gene expression is perturbed in the hemoglobinopathies has pioneered our understanding of the molecular basis of human disease. A key question has been to understand the molecular and cellular basis of the normal switch between the genes encoding the fetal form of hemoglobin (HbF: α2γ2) and those encoding adult hemoglobin (HbA: α2β2).2
The human α- and β-globin subunits are encoded by independent loci (see figure). During fetal life 2 α-globin genes are expressed at the α-globin locus. At the β-globin locus 2 γ-globin genes are expressed during the fetal stage and these are linked to the β-globin gene, which is expressed after birth. Each locus is regulated by distal enhancers (see figure). Research has focused on the developmental switch from γ- to β-globin expression both for its intrinsic biological interest and because preventing or reversing this switch could ameliorate, or even cure, the major hemoglobinopathies, β-thalassemia, and sickle cell disease.3
Initially, the field focused on natural mutations in which the switch from γ- to β-globin expression was fully or partially incomplete. These rare mutations invariably involve deletions, insertions, and point mutations within the β-globin cluster itself. Affected individuals with consistently raised levels (>1%) of HbF have hereditary persistence of fetal hemoglobin (HPFH). As expected, individuals who coinherit one or another form of HPFH with β-thalassemia or sickle cell disease are usually extremely mildly affected.4
During these studies, researchers refined the technologies to accurately and sensitively measure the levels of HbF in the peripheral blood. Individuals with HPFH were found to have very high levels of HbF and, using either immunofluorescence or flow-based assays, researchers found that all (pancellular HPFH) or a substantial proportion (heterocellular HPFH) of the red blood cells (so-called F cells) had readily detectable levels of HbF. It also became clear that the small amount (usually <1%) of HbF found in normal adults was unevenly distributed within a small number of F cells.5
Surveys of healthy blood donors show that the levels of HbF and the numbers of F cells in normal adults vary considerably (∼20-fold). Twin studies have shown that the levels of HbF and F cells have a very high heritability. The continuous distribution of HbF and F cells suggested this variation is controlled by multiple genes and that these are complex traits. By investigating candidate genes, by performing family studies, and via genome-wide association studies (GWASs), researchers identified 3 loci that explain up to 50% of the genetic inheritance of HbF and F cells: the β-globin locus itself (XmnI-HBG2: rs7482144), the BCL11A gene, and the HBS1L-MYB locus. Other loci (eg, LRF, KLF1, Lin28B, and members of the NURD complex) may also influence the levels of HbF and F cells (reviewed in Thein and Menzel6 ).
Acknowledging the complex genetic background controlling HbF and F cells, Khandros et al asked the important question of why individual cells with identical genotypes, and identical combinations of GWAS trait loci, express different levels of HbF? Given the identical genetic background, any satisfactory explanation must involve a probabilistic process in differentiation or gene expression influenced by the known genetic traits. Four explanations come to mind. One is the persistence of fetal-like stem or progenitor cells in adults: this seems unlikely because adult F cells are considerably smaller than bona fide fetal cells and are no more likely to express the fetal i-antigen on their surface than A cells.7 A second explanation is that a small proportion of adult erythroid progenitors exists in a fetal-like transcriptional/epigenetic state. A third explanation is that HbF heterogeneity is caused by small probabilistic changes in gene expression, involving one or more of the known HbF regulators. Finally, the F-cell phenomenon could be explained by alterations in the kinetics of erythroid maturation with premature release of erythroid progenitors that synthesize more HbF than later progenitors.8 Whichever of these explanations is correct, the amount of HbF and the levels of F cells should be explicable in terms the known genetic factors.
To address this, Khandros et al developed protocols to purify and compare F cells and adult cells (A cells). Surprisingly, only the γ-globin genes, and no other causative genes, were differentially expressed at either the RNA or the protein levels. It will be interesting to determine if there is a critical time point at which there are differences in expression of key regulators of F cells. Alternatively, they may involve undetected posttranslational modifications. The difference seen in γ-globin RNA between F and A cells suggests that differential expression of the fetal genes in such cells is controlled by transcription and/or RNA processing rather than translation. Finally, this phenomenon could involve changes in the kinetics of erythropoiesis, in which case the known regulators of F cells must play an important role in this process. Whatever the answer, this classical Popperian approach to the current theories tells us that there is more to find out.
The F-cell phenomenon is not unique. Patients with α-thalassemia who have a common deletion removing both α genes (–SEA/αα) have an excess of β-globin in all erythroid cells, but only 1:1000 of these genetically identical cells have sufficient excess β-globin to form HbH inclusions (HbH cells).4 Similarly, such individuals produce 1% to 2% of red cells in which embryonic ζ-globin chains are detectable (Z cells).9 Looking more broadly in erythropoiesis, there are now many traits that determine, for example, the heritability of red cell size,10 but again genetically identical red cells still show considerable variation. It seems likely that probabilistic variation in any phenotype may occur in genetically identical cells, and, as so often in the past, analysis of globin gene expression in erythropoiesis is leading this interesting area of biomedical research to establish the principles of how such variation occurs and its biological significance.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal