

In this issue of Blood, 1 provide a comprehensive study of the manifestations and clinical course of patients with a particular subtype of Fanconi anemia, FA-B, characterizing the associated genetic variations in the corresponding FANCB gene and their mutational effects and drawing sophisticated genotype-phenotype correlations.
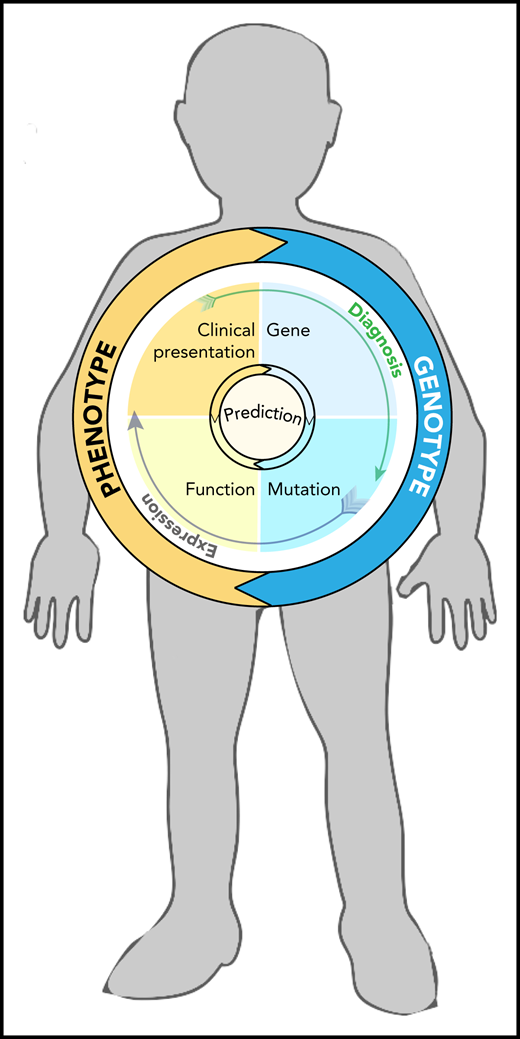
Homunculus with features of Fanconi anemia (short stature, small head, and missing thumb), containing a circle graph that illustrates factors involved in genotype-phenotype relationship and the interdependence of mutation, gene expression, and diagnosis in the process of prediction making (illustrated by arrows).
Homunculus with features of Fanconi anemia (short stature, small head, and missing thumb), containing a circle graph that illustrates factors involved in genotype-phenotype relationship and the interdependence of mutation, gene expression, and diagnosis in the process of prediction making (illustrated by arrows).
From the beginning of the era of molecular genetics, genotype-phenotype analysis has raised hope of better understanding the clinical presentation of a genetic disease, including predicting the natural history and prognosis and, ideally, enabling intervention by precision medicine. In most instances, the results have not lived up to expectations. Mutational diversity still limits our ability to make valid predictions of the course of monogenetic diseases. Classical Mendelian genetics has fostered the illusion that a pathogenic mutation in a specific gene causes monomorphic disruption of gene function and a consistent phenotype. In reality, Mendelian traits can present in many ways with variations in the manifestations of the disease.
Genotype-phenotype correlations often have limitations or are ambiguous. On the clinical side, this may be due to using data extracted from case reports rather than full medical review of patients, inclusion of patients not representative of the entire range of abnormalities, and/or the limited number of patients affected by rare diseases. On the genetic side, the mutational effects of missense, splice, or other variants have not always been studied sufficiently, introducing uncertainty. Regarding inheritance, patients with recessive disorders often have a different pathogenic variant on either allele. Even if either one has been characterized satisfactorily, it is rarely clear what the contribution of each is or their effect in combination. Patients with consanguineous parents carrying a homozygous pathogenic variant in a single gene will have considerable and partly homozygous sequence variation in other genes, making it hard to determine the phenotypic effect.
This is also the situation for the genetic disease Fanconi anemia, which is usually inherited as a recessive trait, apart from 3 reported patients with a dominant-negative de novo mutation in RAD51/FANCR, associated with a Fanconi anemia–like syndrome.2 Fanconi anemia is a heterogeneous disease, with 22 complementation groups and underlying genes reported to date.3 A major function of the Fanconi anemia/BRCA pathway is sensing, removal and repair of DNA interstrand crosslinks. Defects therein result in a disorder with typical yet variable multisystemic congenital malformations.4 . Progressive bone marrow failure with pancytopenia typically presents in the first decade of life.5 Patients experience a high risk of malignancies at an early age, most commonly acute myeloid leukemia and squamous cell carcinoma of the oropharynx and upper gastrointestinal tract and external female genitalia.
Genotype-phenotype analysis in Fanconi anemia patients has been performed in different ways, according to the mutated Fanconi anemia gene (or complementation group), the location of the defect in the pathway, or type of pathogenic variant.6,7 A clear example of a genotype-phenotype relationship in Fanconi anemia patients is the infantile cancer phenotype due to mutations in BRCA2/FANCD1 or PALB2/FANCN. Another correlation is the overlap between Fanconi anemia manifestations and specific sets of abnormalities (associations) such as VACTERL-H (vertebral, anal, cardiac, trachea-esophageal fistula, esophageal atresia, renal, upper limb, and hydrocephalus) and PHENOS (skin pigmentation, small head, small eyes, nervous system involvement, otology, and short stature).7 Within the pathway, the frequency of abnormalities is higher in patients with pathogenic variants in FANCD2 or FANCI, followed by patients with mutations in the downstream Fanconi anemia genes and least in the upstream genes. Regarding the type of variant, a higher proportion of patients with null variants have a more severe phenotype than those with hypomorphic genotypes. Beyond these countable insights, surprisingly few strong genotype-phenotype connections have emerged.
Thus, more clear-cut, state-of-the-art phenotype-genotype correlations are needed urgently. Fanconi anemia patients with the FA-B subtype offer a special opportunity for this, as FANCB is the only Fanconi anemia gene on the X chromosome. Thus, in males, the consequences of a single hemizygous mutation are manifest without the influence of a second allele. Jung et al1 report a cohort of patients from 16 families belonging to the FA-B subtype, a rare Fanconi anemia complementation group that accounts for only ∼2% of patients. The in-depth characterization of 19 patients of this subtype, who were enrolled in the International Fanconi Anemia Registry, is impressive for the number of patients and the detail and quality of the evaluation at both the clinical and molecular levels. There are several lessons that can be learned from this study. On average, FA-B patients showed more severe congenital abnormalities, an earlier onset of hematological features, and a shorter life span than other patients with Fanconi anemia (see figure). FANCB missense mutations attenuate this effect as determined by biochemical and cell-based assays for all reported variants. A high overall proportion of FA-B patients had the VACTERL-H or VACTERL-PLUS association, but these associations were less frequent in patients with FANCB missense mutations. Pathogenic mutations in FANCB are all private (no founders). Approximately one-quarter of pathogenic FANCB variants arise de novo. The variable effects of FANCB missense mutations can be demonstrated by in vitro studies. In the analyzed cohort, Jung et al1 did not detect mosaicism in the hematopoietic system, which could complicate genotype-phenotype relationship and is observed in ∼10% to 15% of Fanconi anemia patients. Mosaicism in an FA-B patient has been reported previously due to an unstable germline FANCB duplication, proven by droplet digital polymerase chain reaction and full-length transcript PacBio sequencing.8 In summary, Jung et al1 reported an exceptionally detailed evaluation of genotype-phenotype correlations. Many results obtained by examination of FA-B patients and pathogenic FANCB variants may be applied to other Fanconi anemia subgroups.
Successful genotype-phenotype correlations are needed to develop combined manifestation- and mutation-specific databases that are currently lacking for most genetic disorders. Even when these databases are generated, other topics still remain challenging. Foremost of these issues is the intricate interplay among mutation, genetic background, environmental factors, and chance events.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal