

Background: Hydroxyurea (HU) is highly clinically beneficial in patients with sickle cell disease (SCD). While HU reduces leukocytosis and inflammation, in general, patients with high levels of fetal hemoglobin (HbF) induction experience the greatest clinical benefit from HU. Despite over 25 years of use in patients with SCD, and intense study, the mechanism by which HU induces HbF has not been elucidated, nor the reasons for the considerable inter-individual variability in the amount of HU-mediated HbF induction. Proposed mechanisms of HbF induction by HU include erythropoietic stress response, production of nitric oxide, or reduction of BCL11A, KLF-1, and MYB, negative regulators of HbF. Various signaling pathways have been implicated in HU-mediated HbF induction, such as the activation of the ERK-1/ERK-2 by erythropoietin; Giα/JNK/Jun; p38/MAPK/CREB1; histone deacetylase (HDAC) and DNA methyl-transferase (DNMT) inhibitor-mediated epigenetic modification of γ-globin expression. To identify new genes and pathways involved in HU activity, we studied the in vivo effects of HU on the gene expression profile of paired samples obtained from the reticulocytes of children with SCD before and after HU treatment.
Methods: 25 paired RNA samples were collected before HU treatment (pre-HU) and at the stable maximum tolerated dose (MTD) from peripheral blood CD71+ reticulocytes of pediatric patients from Texas Children's Hospital. Subjects were 1.2 to 19.1 years of age at the time of pre-HU sample collection (23 HbSS, 2 HbSβ0) and 48% were female. Hematologic indices were extracted from patient's medical record.
RNA sequencing was performed through Illumina platform, paired-end 150 bps, with at least 20 million read in-depth. Quality control of raw sequence data was done with FastQC software. We used Kallisto to determine the compatibility of reads with targets and quantify abundances of transcripts. Then, we used Sleuth to implement statistical algorithms for differential analysis. After validation of results through relative quantity assessment of selected genes by RT-qPCR, the Enrichr web server was used for gene set enrichment (GSE) and pathway analysis.
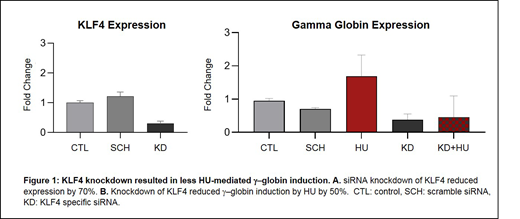
We chose two candidate genes, KLF4 and STAT5A, to analyze functionally based on differential expression, inclusion in enriched pathway analysis, and reported involvement in relevant biological processes, including erythroid maturation or HbF induction. siRNA knockdown of KLF4, and STAT5A in K562 cells was performed by chemical transfection (DharmaFECT) and was verified by RT-qPCR. K562 cells were treated with HU at 50 µM for 24 hours; RNA was extracted and analyzed for γ-globin expression levels by RT-qPCR.
Results and conclusions: We compared our pre-HU and at MTD paired samples using a two‐fold change expression cut‐off and 0.1 FDR multiple comparison correction threshold. Using this threshold, 139 genes had statistically significant differential levels of expression.135 were upregulated and 4 downregulated in response to HU treatment. Pathway analysis primarily grouped the upregulated genes more into the EPO, IL-2, and IL-3 signaling pathways (adjusted p-value<0.05). The functional study using siRNA knockdown experiment revealed that while HU treatment increased γ-globin expression in control cells by 1.7-fold; 70% knockdown of KLF4 resulted in 50% less HU-mediated γ-globin induction compared to control (Figure 1). Scramble (non-targeting) siRNA did not reduce HU induction of γ-globin. Our preliminary results did not show a consistent effect of STAT5A KD on γ-globin induction by HU. We will verify these results in primary erythroid culture, and investigate KLF4 as a potential direct pharmacologic target for HbF induction.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal