

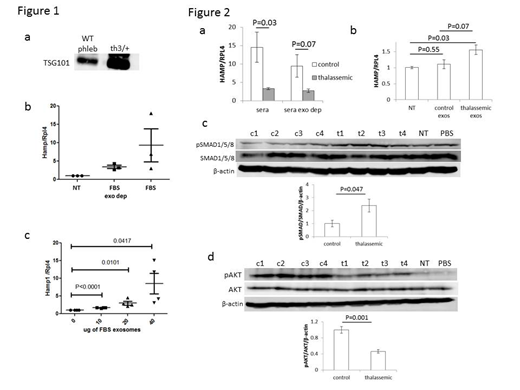
β-thalassemia is characterized by ineffective erythropoiesis and iron overload. Ineffective erythropoiesis causes iron overload by suppressing hepcidin, the main negative regulator of iron absorption and recycling, and is mediated by secretion of erythroferrone from bone marrow cells. Targeted treatment for ineffective erythropoiesis is unavailable. Furthermore, molecular mechanisms involved in ineffective erythropoiesis and the details of how erythropoiesis regulates iron metabolism are incompletely understood. Lastly, while loss of erythroferrone in β-thalassemic mice leads to partial reversal of iron overload [Kautz Blood 2015], erythroferrone ablated mice are still able to suppress hepcidin after phlebotomy [Kautz Nat Med 2014]. These finding provide evidence of additional regulatory crosstalk between erythropoiesis and iron metabolism. We hypothesize that bone-marrow derived exosomes regulate iron metabolism by modulating hepcidin. Exosomes are small extracellular vesicles derived from multi-vesicular bodies forming intraluminal vesicles which fuse with the plasma membrane and are released by many different cell types [Thery Nat Rev Immun 2002]. In light of their capacity for cell-cell communication and modification of the microenvironment, exosomes have been widely studied in multiple diseases [Valadi Nat Cell Bio 2007] despite which, erythropoiesis-derived exosomes and their role in iron metabolism regulation remain unexplored. Our preliminary data demonstrate that phlebotomy in wild type mice results in increased exosome concentration in serum and that exosomes are increased in th3/+ mouse serum (Figure 1a). Furthermore, hepcidin induction by exosome depleted-FBS is decreased relative to FBS (Figure 1b), and exosomes isolated from FBS induce hepcidin in a dose response manner in vitro (Figure 1c). We thus propose to explore the mechanistic relationship between exosomes and hepcidin regulation in β-thalassemia. Serum samples from patients with β-thalassemia major and age / gender matched controls were collected; all patients were treated with iron chelation therapy and all samples were collected immediately prior to transfusion. Exosome fractions were purified and analyzed in patients relative to controls. Although there is no difference in the number of exosomes or mean particle size within the exosomal fraction, exosomal protein content per volume of serum is significantly decreased in patients relative to controls. In addition, the treatment of primary wild type mouse hepatocytes with sera from patients and controls reveals the expected relatively decreased hepcidin induction in β-thalassemic patient sera treated hepatocytes relative to control sera; a similar difference is seen in hepatocytes treated with exosome-depleted sera from patients and controls (Figure 2a). These findings suggest that hepcidin suppression is a consequence of the exosome-free portion of serum from control and β-thalassemic samples. Furthermore, only exosomes derived from β-thalassemic patient sera induces hepcidin expression in primary wild type mouse hepatocyte cultures (Figure 2b). Lastly, exosomes derived from β-thalassemic patient sera do not affect ERK1/2 and STAT3 signaling in primary hepatocytes but increase SMAD1/5/8 (Figure 2c) and decrease AKT signaling (Figure 2d). Taken together, these findings demonstrate that exosomes enhance hepcidin expression via increased SMAD1/5/8 signaling, that increased hepcidin may influence multiple signaling pathways by an autocrine mechanism in response to exosomes, and that exosomes counterbalance hepcidin suppressive substances in the exosome-depleted serum from β-thalassemic samples. Our studies provide novel insights into the important previously unexplored mechanism of hepcidin regulation by exosomes in both physiologic and pathologic states.
Coates:apo pharma: Consultancy, Honoraria, Speakers Bureau; vifor: Consultancy, Honoraria; celgene: Consultancy, Honoraria, Other: steering committee of clinical study; agios pharma: Consultancy, Honoraria. Ginzburg:La Jolla Pharma: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal