

Activated factor VIII (FVIIIa) is an essential cofactor in the intrinsic tenase (Xase) enzyme complex that generates factor Xa and propagates clot formation. The FVIIIa heterotrimer is comprised of a metal ion linked dimer (A1/A3-C1-C2 domains) that is associated with the A2 domain by weak non-covalent interactions. Regulation of FXa formation by the intrinsic Xase enzyme complex occurs by FIXa inhibition and mechanisms contributing to FVIIIa inactivation, including: 1) rapid A2 domain dissociation and 2) activated protein C (APC) cleavage of FVIIIa. While FVIIIa inactivation by APC is considered important, there are surprisingly no in vivo studies documenting the hemostatic role of APC in FVIIIa regulation. Further, published data demonstrate APC cleavage of FVIIIa at physiologic protein concentrations occurs over hours while A2 dissociation occurs rapidly over minutes. Thus, it is thought that the predominant mechanism of FVIIIa inactivation is A2 dissociation and APC likely plays a marginal role in FVIIIa regulation. Additionally, unlike described A2 mutations that enhance dissociation and cause hemophilia A (HA), there is no known disease state attributed to altered FVIIIa cleavage by APC. This is in contrast to FVIII's homologous protein, FVa, whereby resistance to APC cleavage is the most common inherited thrombophilia (FV-Leiden [FVL]). Understanding the physiologically relevant mechanisms of FVIIIa inactivation has immediate clinical applicability for understanding safety considerations in HA therapeutics that bypass FVIIIa regulation (FVIII mimetic antibodies, e.g. emicizumab). Further, as evidenced by successful hemophilia B gene therapy trials using a gain of function FIX variant (FIX-Padua), altering FVIIIa inactivation could be exploited for therapeutic benefit in the setting of gene transfer.
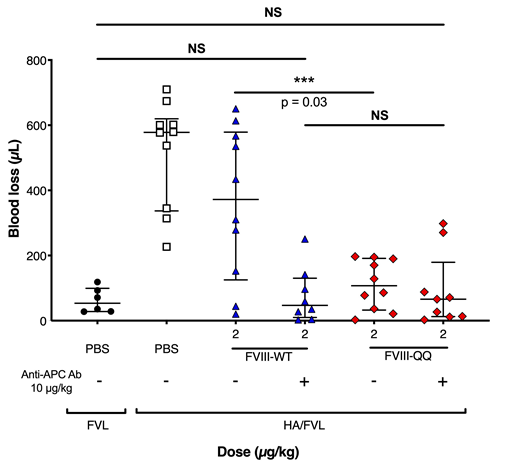
We aimed to determine the in vivo hemostatic role of APC in FVIIIa regulation and pair these studies with purified system analysis. We introduced Arg to Gln mutations at FVIII APC cleavage sites (R336Q and R562Q, herein called FVIII-QQ) on a B-domain deleted FVIII (FVIII-WT) backbone and produced recombinant FVIII-QQ and FVIII-WT. Unlike FVIII-WT, western blot analysis of FVIII-QQ incubated with APC and phospholipids had no evidence of cleavage. Enzyme kinetic studies using purified components demonstrated no appreciable difference in the Km or Vmax for FX within the intrinsic Xase enzyme complex or A2 dissociation of FVIII-QQ relative to FVIII-WT. These data confirmed no unexpected differences in FVIII-QQ relative to FVIII-WT. To begin to evaluate the role of APC in FVIIIa regulation, we measured thrombin generation in murine and human HA plasma reconstituted with FVIII-QQ or FVIII-WT in the presence of increasing APC concentrations. The IC50 of APC was 2-3-fold higher for FVIII-QQ than FVIII-WT. To evaluate the in vivo hemostatic effect of APC in FVIIIa regulation, HA mice were infused with FVIII-QQ or FVIII-WT and evaluated by tail clip injury and 7.5% FeCl3 carotid artery occlusion models. Required doses of FVIII-QQ to normalize blood loss from a tail clip assay and time to vessel occlusion in a FeCl3 assay were 4-5 fold lower than necessary FVIII-WT doses; the superior hemostatic effect of FVIII-QQ supported the physiologic significance of APC in FVIIIa inactivation. To isolate the role of APC in FVIIIa regulation from APC inactivation of FVa, we backcrossed HA mice with FVL mice to create homozygous HA/FVL mice. HA/FVL mice were infused with FVIII-QQ or FVIII-WT and underwent tail clip assay analysis. Doses of FVIII-QQ required to normalize blood loss were again less than FVIII-WT. To further isolate the enhanced hemostatic effect of FVIII-QQ to APC resistance, we performed the tail clip assay in HA/FVL mice infused with FVIII-QQ or FVIII-WT in the presence or absence of MPC1609, an antibody that blocks murine APC function (Xu et al. J Thromb Haemost 2008). In the presence of MPC1609, the same dose of FVIII-WT and FVIII-QQ was required to normalize blood loss (Figure 1). Collectively, our in vitro and in vivo data support the physiologic significance of APC in FVIIIa regulation. To our knowledge these data are the first to demonstrate the in vivo hemostatic effect of APC in FVIIIa inactivation. Our data may be translated to rationally exploit APC regulation of FVIIIa to develop novel HA therapeutics or further delineate safety considerations in therapies that bypass FVIIIa regulation.
Camire:Pfizer: Research Funding. George:University of Pennyslvania: Employment; Pfizer: Consultancy; Avrobio: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal