

Background
RBC transfusion can lead to the production of alloantibodies against RBC antigens and is a clinically significant issue in transfusion medicine. Patients with sickle cell disease have an increased risk for alloimmunization; 30-50% of SS patients have alloantibodies compared to 3-10% of other hospitalized patients. These alloantibodies can cause dangerous hemolytic transfusion reactions and limit the availability of compatible antigen-negative RBC products. This is of particular importance in SS patients, who commonly make alloantibodies against multiple RBC antigens and need regular transfusions to treat their disease. However, mechanisms underlying the increased frequency of alloimmunization in sickle cell patients are poorly understood.
In previous studies, inflammation in the recipient has been shown to promote alloimmunization in both mice and humans. In mouse transfusion models, type 1 interferons (IFNα/β) and Interferon Stimulated Genes (ISGs) have been shown to promote alloimmunization. Other studies have shown that patients with inflammatory autoimmune diseases express an IFNα/β signature, which may contribute to the increased frequency of alloimmunization in these populations. Given the chronic inflammatory state in patients with sickle cell disease, we hypothesize that: SS patients may also have an IFN gene signature that may contribute to the increased frequency of alloimmunization.
Methods
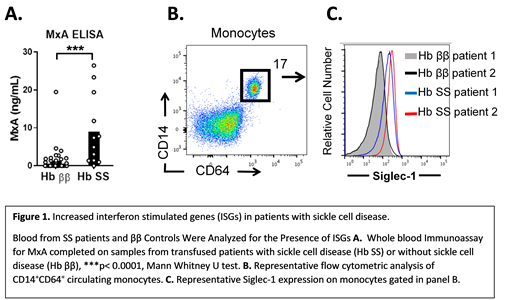
To test this hypothesis, we initially measured the expression of the ISG, myxovirus resistance protein 1 (MxA), in the blood of previously-transfused patients (n=50) with SS disease (SS, n=13) and without SS disease (ββ, n=37) by whole blood immunoassay (ELISA). We then measured expression of another ISG, Siglec-1 (SS n=5, ββ=24), expressed on monocytes by flow cytometric analysis. To determine the degree to which ISG expression correlated with alloimmunization frequency, expression of MxA in non-alloimmunized patients was compared to the expression in patients with 1 or more alloantibodies. Statistical analysis of 2 groups was completed with a Mann-Whitney U test. Significance between 3 groups was determined using a Kruskal-Wallis test with a Dunn's post-test.
Results
SS patients had significantly elevated levels of MxA (mean ± standard error of the mean, SS MxA = 8.98 ng/mL ± 2.46) compared to control patients without SS (MxA = 1.25 ± 0.54, p<0.0001). (Figure 1 A). SS patients also had significantly elevated levels of Siglec-1 on blood monocytes, measured by flow cytometric mean fluorescence intensity (MFI, SS MFI = 132.72 ± 42.9, ββ MFI = 64.9 ± 6.17, p< 0.05). (Figure 1 B,C). For all 50 patients, including SS and ββ control patients, there was a trend toward elevated MxA expression in alloimmunized patients. Patients with 2 or more alloantibodies had significantly elevated MxA (MxA 8.16 ± 2.61), compared to non-alloimmunized transfused patients (MxA = 2.05 ± 1.65, p < 0.01) or patients with only 1 alloantibody (MxA = 1.18 ±0.48, p<0.01). There was no significant difference in MxA levels between patients with 0 and 1 alloantibody. Of the 13 patients with SS disease, only 2 patients lacked alloantibodies. (SS with 1 alloantibody, n=3, SS with 2 or more alloantibodies, n=8). Therefore, a correlation between MxA levels and alloimmunization in SS patients could not be assessed.
Discussion
Factors that contribute to RBC alloimmunization in sickle cell disease are poorly understood. In this study, we found that sickle cell patients had an increase in the expression of ISGs compared to other transfused patients. We also found that MxA levels are increased in patients that have 2 or more alloantibodies compared to patients without alloantibodies. These findings suggest the presence of an IFNα/β gene signature in patients with sickle cell disease. Further studies are needed to determine the relationship between interferon-stimulated responses in sickle cell patients and the increased frequency of alloantibody production.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal