

Background: Targeting the anti-apoptotic BCL2 protein in haematological malignancies has demonstrated significant anti-tumoral activity in a subset of multiple myeloma patients harbouring rearrangements involving the CCND1 and the immunoglobulin heavy chain enhancers (Eμ and α1/2). The mechanisms underlying the dependency of this subgroup of MM patients on BCL2 remains to be elucidated as well as the mechanisms of resistance to BCL2 inhibition with BH3 mimetic venetoclax.
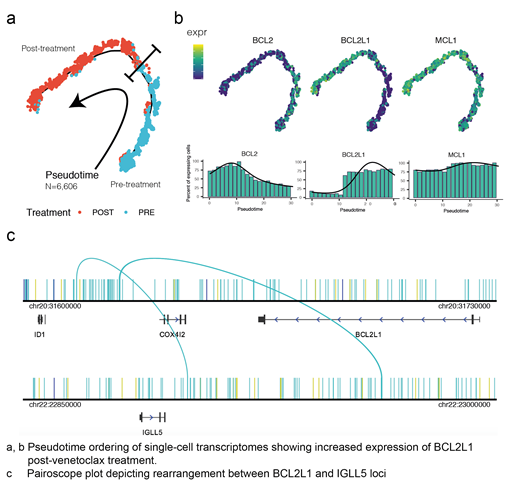
Methods and Results: Sorted bone marrow plasma cells from a cohort of t(11;14) myeloma patients treated with venetoclax were profiled through multi-omics single cell mRNA expression (scRNAseq), copy number profiling (scCNVseq) as well as chromatin accessibility with single cell ATAC-seq. Sequenced reads were aligned to hg38 reference genome. Samples were processed with CellRanger suite v3.0 and downstream analyses were realized with Seurat, Monocle, Signac, and Cicero R packages. Single plasma cells exhibited differential chromatin accessibility landscapes within and across individual patients as well as pre- and post-venetoclax with enrichment of MYC:MAX, RELA, IRF family, RUNX1/3 and ETS motifs. Integration of mRNA and ATAC data revealed a dynamic change of regulatory motifs across individual cell clusters with evidence of selective pressures driven by venetoclax treatment. Similarly mRNA profiling of the apoptotic genes pre- and post-venetoclax exposure showed loss of BCL2 and upregulation of MCL1 and/or BCL2L1 as well as loss of the BH3-only pro-apoptotic genes PMAIP1 and BCC3 in single cell clusters. mRNA levels mirrored open chromatin at the gene bodies and their respective promoter loci consistent with a direct transcriptional regulation. In a patient with several fold upregulation of the BCL2L1 transcript in the post-venetoclax sample (Figure A-B), scATACseq identified a gain in the chromatin accessibility mapping to a genomic region centromeric to BCL2L1 locus on chromosome 20 (chr20:31,617,200-31,619,900). Single cell CNV analysis identified a 5q loss (chr5:142,400,001-156,240,000) mapping to NR3C1 locus explaining with the clinical resistance to dexamethasone. Importantly scCNV also revealed a copy number gain mapping to the same locus with the newly acquired chromatin accessibility on chromosome 20. Mate-pair analysis of the sequencing reads identified the potent IGLL5 B-cell enhancer on chromosome 22 (chr22:22,960,001-22,980,000) as the mate partner juxtaposed the BCL2L1 locus (Figure C). This finding explains the robust upregulation of BCL2L1 mRNA observed in this patient and the shift in BCL2 dependency detected by ex vivo BH3 sensitivity profiling. Of note, while scCNV analysis also depicted a gain in 1q21 (chr1:149,940,001-169,980,001) MCL1 locus at the time of venetoclax resistance the acquisition of t(20,22) shifted the plasma cells dependency to BCL-xL rather than MCL1. This finding was corroborated by the plasma cells ex vivo resistance to dual BCL2 and MCL1 inhibition.
Conclusion: Dynamic single cell epigenome and transcriptome profiling of pre- and post-venetoclax of primary plasma cells identified a de novo translocation driving BCL-xL transcription with the IGLL5 B-cell enhancer. This demonstrates that in addition to canonical TF-promoter regulation, restructuring of immunoglobulin regulatory sequences (i.e., enhancers) can also drive aberrant malignant circuitry endowing resistance to anti-BCL2 agents.
Neri:Celgene, Janssen: Consultancy, Honoraria, Research Funding. Bahlis:Celgene: Consultancy, Honoraria; Janssen: Consultancy, Honoraria; Takeda: Consultancy, Honoraria; Amgen: Consultancy, Honoraria; AbbVie: Consultancy, Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal