

Background
Outcome varies considerably in newly diagnosed myeloma (NDMM), in particular in transplant eligible (TE) patients, ranging from a few years to decades. Autologous stem cell transplant (ASCT) remains the overall aim following induction therapy even with the advent of novel agents. Greater availability of novel treatments allows for complex combination induction and potentially maintenance therapies. Possible long-term unintended effects and affordability will likely make rational treatment stratification for populations in most need desirable, in particular for public health care systems. We report here an extended outcome analysis of 1,064 genetically and clinically characterised patients from the transplant treatment pathway of the NCRI Myeloma XI trial.
Patients
In Myeloma XI patients were randomised to induction therapy with CTD (cyclophosphamide, thalidomide and dexamethasone) or CRD (cyclophosphamide, lenalidomide and dexamethasone) and +/- CVD (cyclophosphamide, bortezomib, dexamethasone) intensification in patients with <VGPR to CRD or CTD. Following protocol amendment patients were randomised between CTD, CRD and KCRD (carfilzomib, cyclophosphamide, lenalidomide, dexamethasone). Following single high dose melphalan conditioned (HD-MEL) ASCT, patients were randomised to maintenance with lenalidomide, lenalidomide+vorinostat, or observation.
Genetic results for the high-risk markers t(4;14), t(14;16), t(14;20), gain(1q) and del(17p) were determined for all patients with sufficient material centrally by qRT-PCR and MLPA (MRC Holland). For a sub-set of patients with insufficient central material, local cytogenetic (FISH) reports were centrally reviewed and only those with a valid result for all risk markers included in this analysis. Differences in PFS, PFS2 and OS were assessed and compared using the log-rank test.
Results
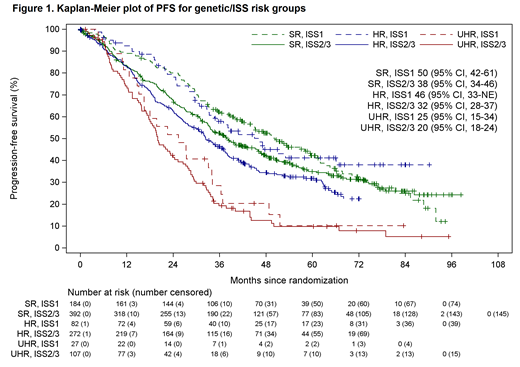
Patient outcome was analysed by combining information on genetic risk defined as standard risk (SR) (no adverse lesions), high-risk (HR) (single adverse lesion: t(4;14), t(14;16), t(14;20), gain(1q) or del(17p)) or ultra-high risk (UHR) (≥2 adverse lesions) and International Staging System (ISS) 1 vs. ≥2. Genetic information was available for all 1064 patients. Baseline clinical characteristics (age, sex, race) were similar between groups, but 47.7% of UHR+ISS≥2 had an IgA paraprotein compared to 27.4% average across all groups. After a median follow up of 68 months (interquartile range 49-83) we found that the two most contrasting risk categories, UHR+ISS2/3 (10.1% patients) and SR+ISS1 (17.3%) were associated with the expected extreme clinical outcomes, with median PFS of 20 months vs. 50 months (P<0.0001) from initial randomisation respectively. Outcomes were similar for UHR+ISS1 with median PFS of 25 months indicating that the main driver to poor prognosis appears to be genetic risk (Figure 1).
Across all genetic risk groups, fewer patients with ISS≥2 vs. those with ISS1 completed intention-to-treat HD-MEL and ASCT. For example, 65.4% of UHR+ISS≥2 completed ASCT vs. 76.1% of SR+ISS1 patients. The main reasons for patients not proceeding to ASCT were 'clinician decision/patient unfit' (43.5%), 'disease progression/death' (26.1%) or 'patient decision' (13%) for UHR+ISS≥2 patients, with fewer 'progression/death' (15%) and a more 'patient decision' in SR+ISS1. Across all groups, more patients were able to undergo ASCT following KCRD induction as opposed to CTD or CRD. This improvement was most notable in the UHR+ISS≥2 group: 82.4% KCRD vs. 58.9% CTD and 67.6% CRD.
Differences in outcomes by risk group were comparable between PFS1 and PFS2. However, for UHR+ISS≥2, median PFS2 was 35 months and OS 43 months (delta=8 months), indicating limited efficacy of current treatments beyond 2nd line for this group. The difference between PFS2 and OS was markedly higher for HR+ISS≥2, with PFS2 52 months and OS 68 months.
Conclusion
Our results indicate the urgent need for improved treatment approaches for ultra-high risk patients, in particular those with UHR+ISS≥2. Our analysis demonstrates that fewer of these patients reach important therapeutic milestones such as ASCT if not effectively treated during induction. These patients are likely to derive the biggest benefit from early stratification and novel treatment approaches, as our data indicate that these patients obtain modest benefit from therapies beyond 2nd line.
Kaiser:Takeda, Janssen, Celgene, Amgen: Honoraria, Other: Travel Expenses; Celgene, Janssen: Research Funding; Abbvie, Celgene, Takeda, Janssen, Amgen, Abbvie, Karyopharm: Consultancy. Jenner:Abbvie, Amgen, Celgene, Novartis, Janssen, Sanofi Genzyme, Takeda: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau. Cairns:Celgene, Amgen, Merck, Takeda: Other: Research Funding to Institution. Pawlyn:Amgen, Celgene, Takeda: Consultancy; Amgen, Janssen, Celgene, Takeda: Other: Travel expenses; Amgen, Celgene, Janssen, Oncopeptides: Honoraria. Paterson:Celgene, Amgen, Merck, Takeda: Other: Research funding to institution. Mottram:Celgene, Amgen, Merck, Takeda: Other: Research Funding to Institution. Davies:Amgen, Celgene, Janssen, Oncopeptides, Roche, Takeda: Membership on an entity's Board of Directors or advisory committees, Other: Consultant/Advisor; Janssen, Celgene: Other: Research Grant, Research Funding. Drayson:Abingdon Health: Consultancy, Equity Ownership. Owen:Janssen: Other: Travel expenses; Celgene, Janssen: Consultancy; Celgene, Janssen: Honoraria; Celgene: Research Funding. Morgan:Celgene Corporation, Janssen: Research Funding; Bristol-Myers Squibb, Celgene Corporation, Takeda: Consultancy, Honoraria; Amgen, Janssen, Takeda, Celgene Corporation: Other: Travel expenses. Cook:Celgene, Janssen-Cilag, Takeda: Honoraria, Research Funding; Janssen, Takeda, Sanofi, Karyopharm, Celgene: Consultancy, Honoraria, Speakers Bureau; Amgen, Bristol-Myers Squib, GlycoMimetics, Seattle Genetics, Sanofi: Honoraria. Jackson:Celgene, Amgen, Roche, Janssen, Sanofi: Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal