

Background
AL amyloidosis is the most common form of systemic amyloidosis, characterized by an associated plasma cell dyscrasia leading to extracellular fibril deposition causing organ dysfunction. In these patients there is a fine balance between treatment toxicities and tolerability due to frailty and presence of multiorgan involvement. This balance is increasingly recognized in the elderly but treatment and outcomes have not been systematically studied, where co-morbidities and frailty may compound morbidity and mortality.
Methods
A retrospective analysis of all patients 70 years or older who were evaluated at the Boston University (BU) Amyloidosis Center from 2000 until 2018 was performed to determine demographics, clinical characteristics, treatments and outcome measures. Kaplan Meier method was used to perform an overall survival (OS) of all patients from the time of diagnosis. Further analysis of OS was performed based on whether treatment was received before or after 2010 (when proteasome inhibitors were incorporated in the treatment algorithm for AL amyloidosis), whether administered treatment regimens consisted of proteasome inhibitors (PI), and whether or not treatment with high dose melphalan and stem cell transplantation (HDM/SCT) was received.
Results
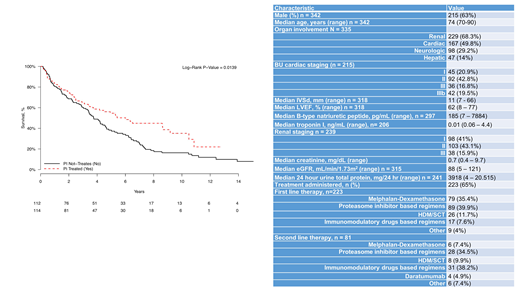
A total of 342 patients with AL amyloidosis who were older than 70 years of age at the time of diagnosis had their initial evaluation at BU Amyloidosis Center from 2000 to 2018. There were 215 (63%) men. The median age was 74 years (range, 70 - 90), and 55 (16%) were older than 80 years of age. The majority of patients were Caucasians (90%). The median number of organs involved by AL amyloidosis was 2 (range, 1- 7). The most common organ involvement was renal in 229 patients (68.3%), followed by cardiac in 167 patients (48.8%), and neurologic in 98 (29.2%). The majority of patients had renal stage II disease and BU cardiac stage II disease. Of the 342 patients, 223 (65%) received systemic treatment, the remainder (35%) received only supportive treatment. The median number of chemotherapy regimens administered was 1 (range 1-5), 116 patients (52%) received one regimen, 81 (36.1%) received two regimens, 35 (15.6%) patients received more than three treatment regimens. Of the 342 patients, 32 (9.4%) received treatment with HDM/SCT. The median age of patients undergoing HDM/SCT was 71 years (range, 70-75).
The median overall survival was 3.4 years (95% CI 2.9-4.1) with a median follow-up of 2.6 years (range, 0.02-15.2). The median OS of patients treated with PI based therapy was 6.0 years vs 3.7 years for those not treated with PI based regimens (p=0.01) (Figure 1). The median OS of patients was 6.8 years for patients treated with HDM/SCT and 4.0 years for those not receiving HDM/SCT (p=0.08). Moreover, the median OS of patients diagnosed prior to 2010 was 3.4 years which is similar to 3.9 years for those diagnosed after 2010 (p=0.07).
Conclusion
In summary, the presentation of elderly patients with systemic AL amyloidosis is similar to that of younger patients in general. There are a higher proportion of patients with advanced cardiac and renal stage disease, perhaps reflecting delay in diagnosis. Compared to supportive care, overall survival in those receiving a PI based therapy is better with respect to survival, although this may reflect selection bias. In addition, HDM/SCT can be offered to highly selected patients with age older than 70 years. Prospective studies in older patients with novel agents with a better toxicity profile and ease of administration may allow a greater proportion of patients to benefit from treatment.
Sloan:Merck: Other: endpoint review commitee; Stemline: Consultancy; Abbvie: Other: Endpoint Review Committee. Sarosiek:Acrotech: Research Funding. Sanchorawala:Proclara: Consultancy, Honoraria; Caelum: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees; Janssen: Research Funding; Prothena: Research Funding; Celgene: Research Funding; Takeda: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal