

Introduction
Primary cutaneous B-cell lymphomas (PCBCLs) arise within the skin without evidence of extracutaneous involvement at the time of diagnosis. The incidence of PCBCL is rare, and PCBCLs are less common than primary cutaneous T-cell lymphomas. There are three subgroups of PCBCL: primary cutaneous marginal zone lymphoma (PCMZL), primary cutaneous follicle-cell lymphoma (PCFCL), and primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL-LT). The 5-year survival rates of the former two subtypes exceed 90%, whereas PCDLBCL-LT is approximately 50% (Wilcox 2015). We describe a rare case of indolent PCMZL transforming into the more aggressive PCDLBCL-LT as well as a review of literature and case series.
Case
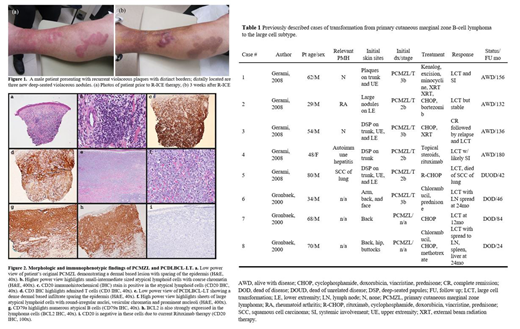
A 62-year-old man with history of PCMZL refractory to external beam radiation and rituximab chemotherapy presented with a two-month history of an enlarging rash and three nodules on his left leg. On physical exam, two violaceous, infiltrative plaques with distinct borders were present on the left leg, resembling his past PCMZL. Three new lesions were described as deep-seated 2 cm erythematous nodules (Fig 1). Histologic review of the new nodules revealed a diffuse, dermal-based atypical lymphoid infiltrate extending from the papillary dermis into the subcutis. The atypical lymphoid cells contained mostly round nuclei, vesicular chromatin, and prominent nucleoli consistent with an immunoblastic and centroblastic morphology (Fig 2). They expressed B-cell markers such as CD79a and CD19, and were negative for CD20 most likely due to prior treatment with rituximab. Immunohistochemistry demonstrated positivity for Bcl-2 and MUM1.
Discussion
Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL-LT) is a rare and aggressive B-cell lymphoma. This subtype is positive for CD20 and CD79, negative for CD5 and CD10, and strongly positive for Bcl-2 and MUM1. It has the gene expression profile of the ABC subtype. A nodule on the lower leg is an ominous sign given this subtype's predilection for lower extremities. This subtype affects the elderly as opposed to the indolent subtypes that arise in middle age (Willemze 2005).
Prognosticating PCDLBCL-LT is challenging, as survival rates vary from 40 to 70% (Willemze 2005). Additionally, the aggressive nature of PCDLBCL-LT confers use of more aggressive, systemic chemotherapy regimens. Rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone (R-CHOP) chemotherapy followed by external beam irradiation is recommended, though no large prospective clinical trials have been performed to date. If lesions remain refractory to R-CHOP, the treatment goal should be redirected to treat as a refractory systemic diffuse large B-cell lymphoma which may require bone marrow transplant or targeted agents with known activity in the ABC subtype such as lenalidomide or ibrutinib.
Transformation from PCMZL to PCDLBCL-LT is a rare event and has only been described in 2 case series (Table 1). In our review of the prior cases, the age group was heterogenous ranging from 29 years-old to 80 years-old. There was no consensus treatment, however, the majority of patients did receive at least CHOP-based regimen. Survival ranged from 24 months to 180 months, showing the heterogeneity of the disease process. In our patient, he underwent R-CHOP but progressed and then received salvage R-ICE with CR. He then underwent bone marrow transplantation but unfortunately had recurrence 36 days post-transplantation, proven PCDLBCL-LT, and he died 161 days following his allogeneic transplant.
Conclusion
Although PCMZL and PCFCL frequently recur despite treatment, transformation to PCDLBCL-LT is unusual. To date, transformation from PCMZL to PCDLBCL-LT has been described rarely in the literature (Table 1). PCMZL is indolent and is treated with external beam radiation therapy or surgical excision with excellent prognosis, the aggressive PCDLBCL-LT subtype has a poor outcome and requires systemic chemotherapy. Because of the vast difference in prognosis and treatment between PCMZL and PCDLBCL-LT, early recognition and exhaustive work-up is beneficial to the patient. Considering this transformation is both rare and diagnostically challenging, clinicians should have heightened concern for new nodular lesions in the setting of recurrent indolent primary cutaneous B-cell lymphomas.
Yazbeck:Celgene: Membership on an entity's Board of Directors or advisory committees; Gilead Sciences: Research Funding; Seattle Genetics: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal