

Chronic lymphocytic leukemia (CLL) has become a paradigm for a cancer that depends on signals from the microenvironment. In lymph nodes (LN), CLL cells receive from surrounding cells proliferative and pro-survival signals, which can protect against chemotherapy and also the Bcl-2 inhibitor venetoclax (VEN). To avoid drug resistance, combination of VEN with the Bruton's tyrosine kinase (BTK) inhibitor ibrutinib (IBR) is currently explored. IBR has the potency to drive CLL cells out of the LN to eventually die by VEN-induced apoptosis. The activation status in the LN likely affects tumor metabolism, though it is yet unclear how, and whether such putative metabolic changes can be linked to VEN resistance. In this study,we aimed to investigate the altered metabolism of CLL cells in the tumor microenvironment (TME), what signals determine these changes, and how to counteract VEN resistance at the metabolic level.
We compared the metabolic status of CLL cells in LN biopsy material and paired peripheral blood (PB) CLL cells. Both higher mitochondrial mass and glucose uptake (flow cytometry) were found in CLL cells derived from LN as compared to PB. To determine which TME signals affect metabolism, we mimicked 3 LN signals: CD40, B cell receptor (BCR) and toll-like receptor (TLR) signaling.Invitrostimulation of PB CLL was followed by mitochondrial mass and glucose uptake (flow cytometry), oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measured on Seahorse XF Analyzer. The datademonstrated that CD40 but surprisingly not BCR or TLR signaling recapitulated the metabolic changes observed in LN cells.
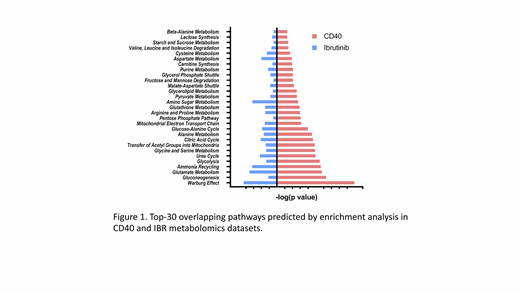
Next, two sets of metabolomics were performed for samples both in vitro and in vivo. Firstly, PB samples of 10 patients were stimulated with/without CD40L for 48 hours and analyzed by mass-spec for metabolic intermediates. In parallel, metabolomics was performed on PB samples from 13 patients sampled before and after 3 months of ibrutinib treatment. The data show various metabolic pathways are activated in the TME, particularly citric acid (TCA) cycle, pyruvate metabolism, glycolysis, and fatty acid metabolism. Apart from these general changes, the highest-ranking shifts in metabolites point to involvement of amino acids to fuel the TCA cycle, which in turn drives oxidative phosphorylation (OXPHOS). Overall, CD40 signaling results in increased glycolysis, but more dominantly, increased OXPHOS, while IBR in fact has opposing effects (Figure 1), indicating that TME-driven metabolic alterations are mitigated by IBR treatment.
In order to link the altered metabolic state to VEN resistance, PB-derived cells were treated with OXPHOS inhibitors during CD40 stimulation. Remarkably, OXPHOS inhibition by oligomycin (ATP synthase inhibitor) did not affect CLL activation, yet counteracted strongly for VEN resistance. Of several BCL-2 family members induced by CD40 ligation, both anti-apoptotic MCL-1 and BCL-XL were downregulated by oligomycin. These data suggest that OXPHOS inhibition affects CD40 signals to counteract VEN resistance.
In conclusion, metabolic changes of CLL in LN are recapitulated by CD40 signal, while IBR treatment shows opposite effects, together providing indirect insight into the LN metabolism. In LN, most of the key metabolic pathways are enhanced, particularly OXPHOS. Finally, we found OXPHOS inhibition reverses CLL VEN resistance. Our findings link CLL metabolism in LN microenvironment to VEN resistance, and may provide novel clues for therapeutic targeting in the treatment of VEN resistance patients.
Forconi:Roche: Honoraria; Gilead Sciences: Research Funding; Abbvie: Consultancy, Honoraria, Other: Travel, Accommodations, Expenses, Speakers Bureau; Menarini: Consultancy; Novartis: Honoraria; Janssen-Cilag: Consultancy, Honoraria, Other: Travel, Accommodations, Expenses, Speakers Bureau. van der Windt:Genmab: Employment. Eldering:Celgene: Research Funding; Janssen Pharmaceutical Companies: Research Funding; Roche: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal