

Background and Aim: The entity defined by the WHO 2017 classification as myeloid neoplasms with germinal predisposition without preexisting disorder or organ dysfunction is particularly interesting within myelodysplastic syndromes (MDS) for three main reasons: i) in myeloid disease derived from congenital bone marrow failure, therapeutic strategies (e.g., type of conditioning regimen) are defined; it is not the case within the group of young patients with MDS harbouring germinal variants, actually a group candidate for allogeneic transplantation of hematopoietic progenitors, ii) its incidence exceeds the cases secondary to congenital bone marrow failure, and iii) the implications of genetic counseling to patients and relatives are yet to be defined and have not been addressed at the time of diagnosis of MDS.
Methods: Whole exome sequencing (WES) was performed on 118 tumour and 73 paired germinal DNA from patients of 16 Spanish Group of MDS (GESMD) centers, diagnosed with de novo MDS between 16-60 years old without previous organ dysfunction. WES libraries were sequenced on a HiSeq4000-NovaSeq6000-Illumina platform, mean number of reads per sample was 144,429,985 with a Phred Quality Score >30 in 94% of bases and a 100x average depth. To identify potential germline-predisposing mutations, a selection tool incorporating 279 genes associated with cause or predisposition to bone marrow failure or cancer was implemented. The analysis of the variants was carried out by means of an in house pipeline: filtering out intronic, synonymous, and those variants with minor allele frequency (MAF) in the general population >1% (ExAC, 1000 Genomes-phase3, TOPmed), and requiring the presence both in tumour and germline DNA with a VAF>37%. In 45 cases without germline material the last requirement was substituted by not being reported as somatic in COSMIC in any cancer. To determine pathogenicity we followed conservative criteria: a CADD Phred score ≥20 and to be considered deleterious in, at least, three out of six used algorithm.
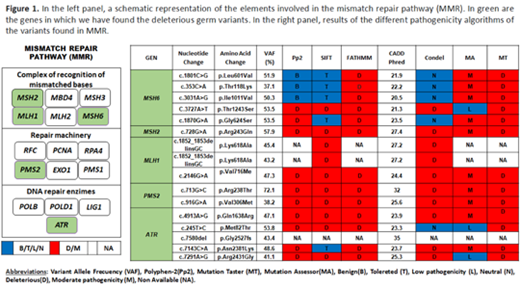
Results: In 118 patients, the median age at diagnosis was 47 years with the following WHO 2017 diagnoses: 12% MDS-SLD, 9% MDS-RS, 34% MDS-MLD, 30% MDS-EB, 3% MDS-del(5q), 1%MDS-U, 11% CMML. We found deleterious/pathogenic germ variants in 68 of 118 patients. Strikingly, we found a higher frequency than expected, for this specific subset, in genes not yet considered in the category of myeloid neoplasms with germline predisposition: MSH6 (n=5;4.2%), ATR (n=5;4.2%), ERCC6L2 (n=4;3.4%), PMS2 (n=2; 1.6%), MLH1 (n=3;3.5%), HCLS1 (n=2;1.7%), ITGB3 (n=2;1.7%), LYST (n=4; 3.4%), SAMD9 (n=1;0.8%), MSH2(n=1;0.8%). In genes already considered in the category of myeloid neoplasms with germline predisposition: MPL (n=2;1.7%), DDX41 (n= 2;1.7%), RUNX1 (n=1; 0.8%), ANKRD26 (n=1;0.8%). We also detected 10 cases with deleterious germline variants in genes related to Fanconi anemia (BRIP1, FANCC, FANCD2, FANCG, FANCM, SLX4, XRCC2, RAD51C and BRCA2), 2 cases with a germline variant in a Shwachman-Diamond gene DNAJC21 (1.7%), and 3 cases with germline variant in a telomere biology gene (RTEL1, CTC1 and TERT). We then focused on the characterization of the variants found in 5 genes not considered to date as predisposing to MDS: MSH6, MSH2, MLH1, ATR and PMS2 involved in the instability of microsatellites and whose alteration determines genomic instability and predisposition to a different number of solid tumors. The frequency of patients carrying these variants (13.5%) is much higher than the frequency of DDX41 (1.7%) or CEBPA (not found in our cohort), the two genes currently considered in the category with germline predisposition without a preexisting disorder or organ dysfunction. The sixteen patients carrying these mutations were characterized by a median age of 47 (16-60) years with the following diagnoses, 33% MDS-MLD, 27% MDS-EB1 and 40% CMML, presenting up to 40% with decrease cellularity in the bone marrow.
Conclusions: We describe, for the first time, a high frequency of germinal variants in genes that drive genomic instability by modulating the microsatellite pathway, in young adults diagnosed with MDS without previous organ dysfunction. Their frequency and high pathogenicity index warrant functional validation experiments and pose them as potential candidates to be included in future classifications and to be considered in clinical, therapeutic and genetic counseling strategies.
Díez-Campelo:Novartis: Consultancy, Membership on an entity's Board of Directors or advisory committees, Research Funding; Celgene Corporation: Consultancy, Membership on an entity's Board of Directors or advisory committees, Research Funding. Sanz:Celgene: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Helsinn Healthcare: Membership on an entity's Board of Directors or advisory committees, Research Funding; Onconova: Membership on an entity's Board of Directors or advisory committees, Research Funding; Hoffman - La Roche: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Amgen: Membership on an entity's Board of Directors or advisory committees, Research Funding; Boehringer-Ingelheim: Membership on an entity's Board of Directors or advisory committees; AbbVie: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Janssen - Cilag: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Novartis: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding. Jerez:Novartis: Honoraria; Celgene: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal