

Background:
Sickle cell disease (SCD) is a monogenetic inherited red cell disorder with pleomorphic clinical manifestations. Hemoglobin F (HbF) concentration is the major genetic modifier of clinical expression and levels between 10% and 20% have been found to improve survival and decrease vaso-occlusive complications (VOCs). As survival for patients with SCD has increased dramatically in the past 20 years, we hypothesized that SCD-related complications may increase with age, even in "high F" individuals. Recent data suggest that individuals with the Arab-Indian haplotype and/or HbS-δβ thalassemia, who have HbF of 15% to 25%, still suffer from VOCs. SCD patients with HBS and deletional hereditary persistence of fetal hemoglobin (HPFH) have high levels of HbF (>30%) in a pancellular distribution, and absence of symptoms as suggested by prior studies. Here, we report on a cohort of patients with HbSS/Sβ0, with HbF >14%, either hydroxyurea naïve or treated for ≤ 6 months.
Methods:
We report on a cohort of 11 adults with SCD and HbF levels > 14%, who had never been on hydroxyurea, or were treated for ≤6 months. Charts were reviewed for VOCs, opioid therapy, acute chest syndrome (ACS), avascular necrosis (AVN), retinopathy, pneumonia, leg ulcers, splenomegaly/infarction, stroke, pulmonary hypertension, cardiomyopathy/infarction, chronic kidney disease, priapism and miscarriage. All events were confirmed by supportive laboratory or imaging data. HbF levels for each patient are represented as lifetime average values. In 10/11 patients, cellular distribution of HbF by flow cytometry (CytoFlex, Beckman Coulter, CA) was performed following fixation, permeabilization, and incubation of red blood cells with a monoclonal antibody against Hb-F, directly conjugated with FITC (Cat # MHFH01, Life Technologies, MD). Beta globin dosage genetic analysis was performed on a subset of patients (8/11) which used multiplex-polymerase chain amplification of fragments specific for portions of the epsilon, gammaG, gammaA, delta, and beta globin genes, which is commonly associated with deletional variant of HPFH (Quest Diagnostics, VA).
Results:
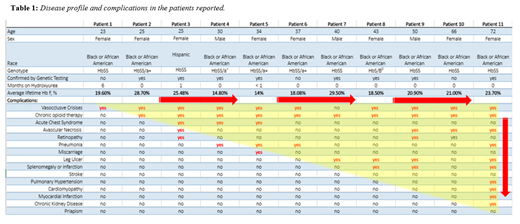
Our cohort consisted of 11 patients, 3 males, age range 23 -72 years, one of which (age 72) is deceased. Genotypes included 10 HbSS, four with alpha thalassemia trait, and one HbS/β0. Mean HbF levels ranged from 14 % to 29.50%, SD ± 4.76. Table 1 shows patients ordered in ascending age with clinical complications for each patient. Notably, all patients experienced VOCs regardless of age or HbF level, 91% were prescribed opioids, 46% of whom were on chronic opioid therapy, 40 % had leg ulcers 36% had AVN and 27% had ACS . The number of complications per patient increased with age, despite maintenance of high HbF. Available flow cytometry and genetic data revealed heterocellular distribution of HbF in 8 patients, and pancellular distribution in 2 patients: patient (#2) and patient (#10). Patient (#2) is positive for deletional HPFH with genetic confirmation of heterozygous positive deletion in the beta globin gene cluster. Genetic data is not available for patient (#10) at this time. Interestingly, both patients still exhibited VOCs, complicated by yearly hospitalizations, required intermittent opioid therapy and with age, developed AVN. Laboratory data are notable for minimal levels of hemolysis.
Discussion:
Most individuals with SCD living in high resources countries now reach adulthood. Therefore the disease profile that was previously studied and reported mostly in the pediatric population is evolving. High levels of HbF appear to delay the onset of complications, but do not completely prevent them, perhaps due to a heterocellular distribution of HbF or non-deletional HPFH. In our cohort, two patients with confirmed pancellular distribution of HbF, one of which with confirmed deletional HPFH, were not fully protected from the clinical complications of SCD, suggesting alternative pathogenic mechanisms for VOCs may be at play. Therefore, individuals with HbF concentrations of as high as ~30 % may still suffer from pain, hospitalizations for VOC's, and vasculopathic changes, such as leg ulcers. The number of complications increases with age despite maintenance of high HbF. Therefore, additional therapy and close vigilance is necessary when treating sickle cell patients with high levels of HbF, especially with increasing age.
Reyes Gil:Albert Einstein College of Medicine: Patents & Royalties: Patent application pending for NETs AI software. Ogu:Vertex Pharmaceuticals: Consultancy. Minniti:Doris Duke Foundation: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal