

Sickle cell disease (SCD) is characterized by increased oxidative stress. Sources of oxidative stress include (1) intermittent vascular occlusion resulting in hypoxia-reoxygenation induced activation of prooxidant enzymes such as xanthine oxidase and NADPH oxidase, (2) increased fragility of red blood cell membranes causing hemolysis and release of free hemoglobin and heme which produce reactive oxygen species (ROS) through Fenton chemistry, and (3) respiratory chain leaked electrons reacting with oxygen to form superoxide. In SCD, the antioxidant defense system has been shown to be insufficient in its response to increased ROS production. In particular, peripheral blood expression of the mitochondrial targeted antioxidant superoxide dismutase 2 (SOD2) is decreased in SCD patients relative to non-sickle controls. We hypothesize that depletion of SOD2 may modify the progression of sickle pathology.
In order to test our hypothesis we (Aim 1) genotyped 410 SCD patients from the Treatment of Pulmonary Hypertension and Sickle Cell Disease with Sildenafil Therapy (walk-PHaSST) study for a common polymorphism of SOD2 (rs4880), valine to alanine on the 16thamino acid (V16A), and investigated whether or not the polymorphism is associated with clinical indicators of endothelial dysfunction. (Aim 2) In primary human pulmonary arterial endothelial cells (hPAECs) we utilized an siRNA mediated knockdown of SOD2 (SOD2 KD) in order to examine its role in maintaining endothelial cell permeability. Permeability was measured by using electric-cell substrate impedance sensing (ECIS).
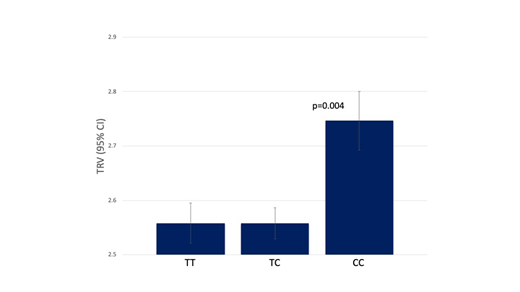
In Aim 1, among the 410 SC anemia patients 129 (31%), 64 (16%) and 217 (53%) were homozygotes for the common valine allele (TT), homozygotes for the variant alanine allele (CC) and heterozygotes (TC), respectively. We examined association of each genotype with the following clinical parameters: history of pulmonary embolism, systolic blood pressure (SBP), pulse blood pressure, hemoglobin, mean corpuscular value, reticulocyte count, white blood cell count, platelet count, tricuspid regurgitant velocity (TRV), left mass index, right atrial area, left ventricle, right ventricular area at systolic, creatinine, six minute walk distance (6MWD). We found that homozygotes of alanine variant (CC) had higher systolic blood pressure (p=0.011), higher TRV (p=0.004), larger right ventricular area at systolic (p=0.023), as well as shorter 6MWD (p=0.006). All four of these clinical parameters are strong indicators of vasculopathy and endothelial damage.
Based on our clinical findings, we extended our studies to isolated endothelial cells to define the role of SOD2 in endothelial cell permeability. In Aim 2, we used ECIS in order to measure resistance in SOD2 KD hPAECs. Decreased resistance as measured by ECIS has been shown to be an indicator of increased permeability. We found that SOD2 KD hPAECs had decreased baseline resistance as compared to hPAECs treated with a non-targeting siRNA sequence (siNT). This data supports that SOD2 plays a role in maintaining endothelial cell barrier function. We also, investigated whether free hemin would further accentuate endothelial barrier dysfunction in SOD2 KD cells. We serum starved siNT and siSOD2 KD hPAECs for four hours before treating with 2 mM hemin and measuring resistance. We found that after four hours of 2 mM hemin treatment, there was no further reduction of resistance in SOD2 KD hPAECs as compared to siNT hPAECs.
In conclusion, we have found that in SCD patients SOD2 V16A is associated with clinically significant indicators of endothelial dysfunction and SOD2 is essential for the maintenance of endothelial cell barriers in hPAECs. Future directions will be aimed at further investigating the SOD2 V16A polymorphism, specifically we are interested in whether the polymorphism plays a role in barrier function. We will also investigate the pathways by which SOD2 depletion mediates endothelial cell barrier function. Taken together, our preliminary findings suggest that SOD2 functions as an essential mediator of endothelial function in SCD and thus can be used as a target for future SCD therapeutics.
Straub:Bayer Pharmaceuticals: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal