

Background: Clonal heterogeneity is a known issue in multiple myeloma (MM) and the emergence of drug resistant clones is responsible for the incurability of the disease. Multiple studies of bulk CD138+ bone marrow samples have attempted to stratify MM patients into smaller, more distinct, patient risk groups based on molecular phenotypes. Recently, single cell RNA sequencing (scRNA-seq) technology has been applied in MM to identify cell clones. This leads to a new question: can we classify patients with scRNA-seq data guided by previously defined subtypes, and how do the single cell results correspond with the classification?
Methods: We developed a novel, deep transfer learning framework to predict MM patient subtypes in patients with scRNA-seq based on patient classifications from microarray data. While the problem of scRNA-seq batch corrections has been intensively studied using transfer learning, there has been less work on similar comparisons between scRNA-seq and patient-level data. To address this issue, we utilized domain adaptation, a specific transfer learning approach, to combine scRNA-seq profiles and patient-level microarray data using a multitask learning framework.
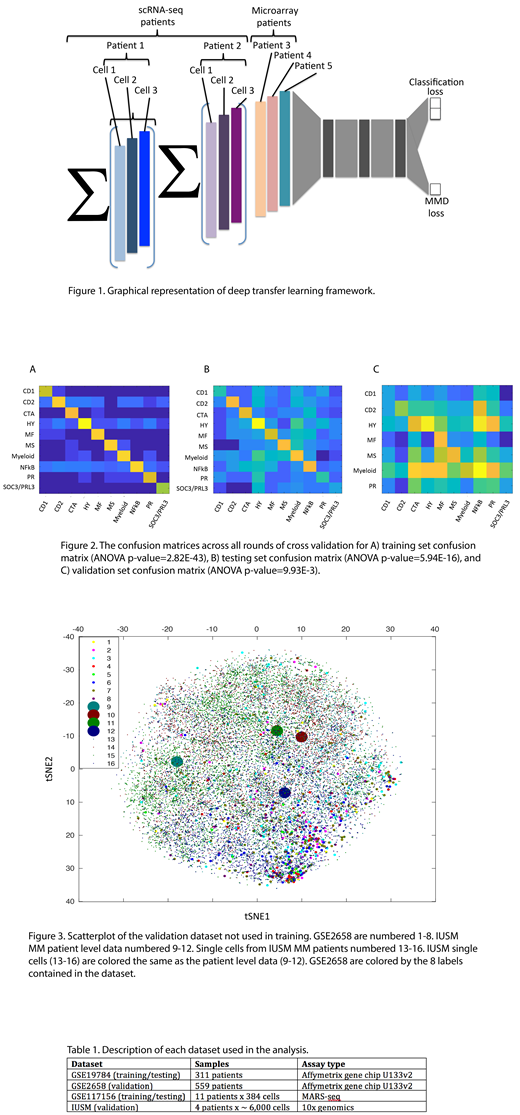
Figure 1 illustrates our computational framework. Its aim is to classify both cells and patients (with scRNA-seq data) according to patient level classifications derived from previous gene expression profiling studies for MM. Specifically, we adopted the 10-subtype classifications derived from microarray data1. Patients with scRNA-seq were summarized into a single vector by averaging gene counts across all the cells. Gene expression profiling data (including scRNA-seq and microarray) for MM patients from multiple studies were input into the transfer learning network consisting of 5 hidden layers. The last hidden layer was used to calculate the maximum mean discrepancy (MMD) between the patients from scRNA-seq and microarray to integrate the datasets.
The datasets in this study are summarized in Table 1. Two microarray datasets (GSE19784, GSE2658) and one scRNA-seq dataset (GSE117156) were obtained from NCBI Gene Expression Omnibus. IUSM data were locally generated. One microarray and one scRNA-seq dataset were used in training and testing. GSE19784 was split into 80% training and 20% testing. GSE117156, due to the smaller sample size (11 patients), was split into 90% training and 10% testing. We ran 20 rounds of random cross validation using TensorFlow on a GTX1080 GPU. The expression profiles of patients and single cells from all datasets (GSE19784, GSE117156, GSE2658, IUSM) were input into the trained model after each round of cross validation to produce low-dimensional representations and predictions for each training, testing, and validation sample.
Results: We found that our model was able to identify signals in the data based on expression profiles from patient-level and single cell data. The patient classification labels can be consistently reproduced in a held-out test set of patients as well as in a validation cohort of microarray data from 559 MM patients (GSE2658) and scRNA-seq from 4 MM patients from IUSM (Figure 2). These results show that the model can learn the subtypes across multiple datasets and platforms.
The 4 IUSM patients tended to cluster similarly to their individual CD138+ cells after training, while GSE2658 patients still maintained some separation between MM subtype clusters (Figure 3). The single cells from our cohort of 4 patients did not necessarily classify to the same subtype as their patient.
Conclusions: We found that a domain adaptive classifier can be trained across scRNA-seq and bulk gene expression profiling data from MM patients to integrate data and transfer knowledge. These models showed that single cells within a patient do not necessarily match the patient level molecular characteristics. Not surprisingly, similar results have been found in other cancer types2. As our novel framework is further refined and more patients are sequenced, we expect more unique insights into both inter- and intra-tumor MM heterogeneity.
References:
1. Broyl A, Hose D, Lokhorst H, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood. 2010;116(14):2543-2553.
2. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396-1401.
Abonour:Celgene: Consultancy, Research Funding; BMS: Consultancy; Takeda: Consultancy, Research Funding; Janssen: Consultancy, Research Funding. Roodman:Amgen: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal