

Background
Acute myeloid leukemia (AML) is a molecularly and clinically heterogeneous hematological malignancy. Chemotherapy resistance is common, and relapse is the major cause of treatment failure. Investigation of new molecularly-targeted and immunomodulating agents therefore remains a high priority. Interferon (IFN)-γ regulates inflammatory responses and tumor immunosurveillance. Prolonged IFN-γ signaling may promote immune-independent resistance to genotoxic anti-cancer therapies. Herein, we aimed to establish whether tipifarnib, a farnesyl-transferase and CXCL12/CXCR4 pathway inhibitor currently being tested in phase I/II clinical trials in individuals with myeloid malignancies, modulates IFN-γ signaling.
Methods
We previously defined an IFN-γ-responsiveness gene signature in AML cell lines (Vadakekolathu J, et al. Blood 2017; 130: 3945A). Herein, we employed targeted gene expression profiling for the high-dimensional analysis of canonical signaling pathways and their modulation by tipifarnib in Kasumi-1 AML cells [AML with t(8;21)] and KG1 AML cells (leukemia stem cell [LSC]-like AML). AML cells were either incubated with 100 nM or 500 nM tipifarnib for 6 hours, or left untreated, prior to in vitro challenge with 10 ng/mL IFN-γ for 24 hours. RNA (approximately 100 ng per sample) was incubated with a reporter and capture probe mix for hybridization (mRNA Pan-Cancer Pathways Panel; NanoString Technologies, Seattle, USA). Transcript counts were analyzed on the nCounter FLEX analysis system. The nSolver™ software package and nSolver Advanced Analysis module were used for quality controls. The captured transcript counts were normalized to the geometric mean of the housekeeping reference genes included in the assay and the code set's internal positive controls. Differentially expressed genes were assessed in silico for correlations with clinical-biological disease characteristics and potential prognostic value in The Cancer Genome Atlas (TCGA)-AML cases (162 sequenced AML samples with putative copy-number alterations, mutations and mRNA expression z-scores [threshold±2.0]).
Results
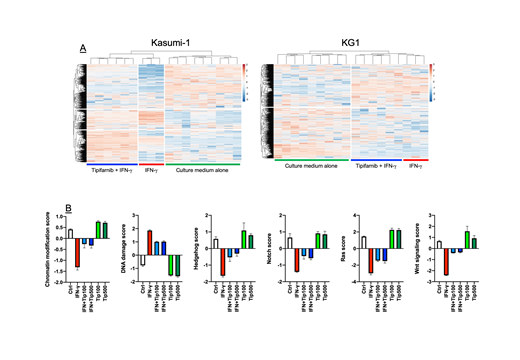
Unsupervised hierarchical clustering of mRNA expression identified a set of IFN-γ-stimulated genes (ISGs) that were up-regulated (>2.0 fold-change compared with baseline) in Kasumi-1 cells, but not in KG1 cells, in response to IFN-γ treatment. When assessed in silico for prognostic power in TCGA-AML patients treated with curative intent on a '7+3' chemotherapy backbone, ISGs in our experimentally-derived signature (SOCS1, FAS, ETV7, IL15, SOCS3, PIM1, TNFSF10, STAT1, PLAT, PRDM1, CASP7, BCL2A1) were either up-regulated, amplified, deleted or mutated in 59 (36%) of queried samples. Patients with abnormalities of ISGs experienced worse clinical outcomes, as indicated by shorter relapse-free-survival (RFS; median 12.1 versus 24.2 months in AML patients without ISG abnormalities, p=0.041) and shorter overall survival (OS; median 11.8 versus 24.8 months in AML patients without ISG abnormalities, p=0.036). Interestingly, abnormalities in ISGs significantly correlated with TP53 mutations (log ratio= 1.65; p=0.026), an established adverse prognosticator in AML. Pre-treatment with tipifarnib at either 100 nM or 500 nM concentration attenuated IFN-γ-induced changes in gene expression in Kasumi-1 AML cells (Fig. 1A), including the down-regulation of Wnt, Ras, Hedgehog and Notch signaling and the up-regulation of genes implicated in DNA damage response (Fig. 1B). In contrast, tipifarnib exerted no effects on cancer-associated canonical pathways in IFN-γ-unresponsive KG1 cells (Fig. 1A).
Conclusions
Our study shows that an experimentally-derived IFN-γ responsiveness gene signature correlates with poor clinical outcomes in AML and suggests that tipifarnib might attenuate IFN-γ signaling, potentially affecting AML susceptibility to DNA damage induced by chemotherapeutic agents.
Grant support: Qatar National Research Fund (#NPRP8-2297-3-494) and Kura Oncology, San Diego, CA, USA.
Rutella:MacroGenics, Inc.: Research Funding; NanoString Technologies, Inc.: Research Funding; Kura Oncology: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal