

Background. Inherited factor VII (FVII) and factor X (FX) deficiencies are rare coagulopathies with heterogeneous genetic background. Combined FVII-FX deficiency was also described.
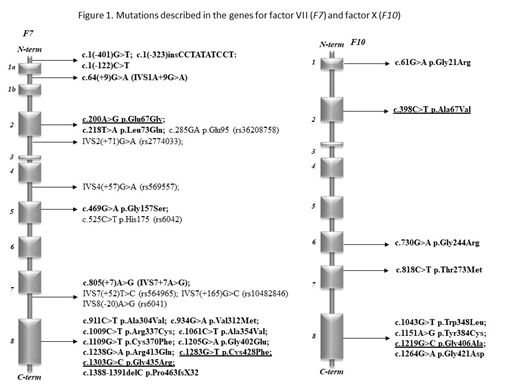
However, genotype-laboratory and clinical phenotype correlations have rarely been studied, in particular in pediatric patients. Methods. We investigated adult and pediatric patients with FVII and FX deficiency in a tertiary care center and analyzed the genotype-phenotype relationship and consequences of novel mutations by in silico methods. The study was approved by the National Ethics Committee (No. 58523-4/2017/EKU) and conducted according to the Declaration of Helsinki. All patient-related data were anonymized and represent results of routine clinical and laboratory investigations so as to establish the exact diagnoses and the optimal treatment of patients. Activity levels of coagulation factors including FVII and FX were determined by PT-based one-stage clotting assays. FVII antigen concentration (FVII:Ag) was measured by ELISA. The whole coding region, the exon-intron boundaries and the promoter region of F10 and F7 were amplified by PCR under standard conditions. Purified PCR products were directly sequenced with the Big Dye Terminator Cycle Sequencing and resolved on an automated ABI PRISM 3130 DNA sequencer. Sequencing data were analyzed by the Sequencing Analysis 5.4 software. UniProtKB/Swiss-Prot database was used for investigating the conservative or un-conservative nature of the mutated amino acids and surroundings in case of novel missense variants. The novel missense variants found in F7 or F10 genes were analyzed by free bioinformatics tools, MutPred2, PolyPhen2, PredictSNP, and Mutation Taster for pathogenicity. Results. Between 2003 and 2018, 18 individuals with FX deficiency (1-41 yrs) and 31 patients with FVII deficiency (5-85 yrs) were identified. Sixteen different pathogenic mutations were found in F7. Novel variants were p.Glu67Gly, p.Cys428Phe and p.Gly435Arg. Most common combinations were p.Arg413Gln with p.Ala354Val and p.Arg413Gln with IVS1A+9G>A. Eight mutations, including four novel ones (p.Ala67Val, p.Gly406Ala, p.Ther273Met and p.Trp348Leu), were found in F10. p.Gly406Ala and p.Thr273Met proved pathogenic by all prediction tools and 2/4 methods suggested pathogenicity of p.Ala67Val and p.Trp348Leu. Similar mutation pattern was observed in four unrelated families with combined FX and FVII deficiency suggesting founder effect. Our cases supported the possibility of independent occurrence of small genetic alterations, like missense mutations and small deletions, affecting both genes. Regarding genotype-phenotype relationships, FVII activity values in patients with symptoms (most frequently epistaxis, menorrhagia and bleeding upon minor surgery) were between undetectable and 48 U/dL. Interestingly, patients without any bleeding showed a wide range of FVII activity (between undetectable and 61 U/dL). There were no associations between F7 genotype, FVII activity, and clinical symptoms in our population. There were 8 surgical interventions performed in rFVIIa coverage with excellent bleeding control. All patients with FX activity <1U/dL were homozygous or compound heterozygous for F10 mutations and had severe bleeding manifestation symptoms requiring PCC prophylaxis. The heterozygous carriers had FX activity levels between 45 and 67% and only one patient with isolated mild FX deficiency had bleeding symptoms. Six persons belonging to four families had combined FX and FVII deficiency. All of them were heterozygous for FX deficiency having FX activity between 47 and 66%. One patient had FVII activity below 1% caused by homozygous mutations in F7, which was associated with umbilical stump bleeding. Conclusions. There are no large clinical studies available in FX and in combined FVII-FX deficiencies. Similarly, no large studies investigated the genotype- clinical and laboratory phenotype relationships in patients with FVII deficiency. Consequences of mutations in the F7 and F10 gene should be investigated at the molecular level. As novel treatment strategies are rapidly emerging in the field of rare bleeding disorders, clinical, laboratory and molecular investigations of larger cohorts, as the present one, are deemed important to guide optimal management of patients.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal