

Introduction: Several studies have attempted to describe the characteristics associated with large granular lymphocytosis (LGL) following allogeneic hematopoietic stem cell transplantation (allo-HSCT) and its clinical significance. However the clinical features of LGL lymphocytosis in the allo-HSCT setting is still sparse. The current study represents a detailed review of 667 patients transplanted in a single center with the objective to define the incidence of LGL lymphocytosis, to identify associations with transplant-related clinical parameters and to assess the impact on transplant related outcomes.
Patients and Methods: During a 14-year follow up period (2005-2017) in this unicentric cohort study, we identified 19 patients (2.8%) with a significant LGL lymphocytosis, among 667 consecutive adult patients who underwent allo-HSCT. LGL lymphocytosis was defined as the presence of at least two of the following criteria: (1) sustained lymphocytosis above 3.0x109/L observed in at least three consecutive determinations over a time frame of 2-3 months, (2) predominance (that is, >30%) of LGLs in the peripheral blood, (3) confirmation of clonality by T-cell receptor analysis using PCR. Flow cytometry analyses were performed using the flow cytometry system FACSCalibur (BD Biosciences, San Jose, CA). The immunophenotyping of the lymphocytes included the following antibody panel: CD2, CD3, CD4, CD5, CD7, CD8, CD16, CD25, CD30, CD56, CD57, HLA-DR, TCRab, and TCRgd. T-LGL expansion was defined as an abnormal T cell population type CD31, CD81, or CD41, with expression of at least 1 of the NK markers (CD16, CD57, or CD56), and with presence of LGLs in peripheral blood films.
Results: A total of 19 (Female/ male: 10 [52.6 %]/ 9 [47.4 %]) patients included into the study met the morphological criteria for LGL lymphocytosis. The median age of the patients was 46 years (range, 18- 62 years). The majority of the patients (64.7 %) had the diagnosis of acute myeloid leukemia. The stem cell source was peripheral blood stem cells (PBSC) in 15 patients (88.2 %) and most of the patients underwent an allo-HSCT with a MAC (n= 13) regimen at a median of 25.1 months from allo-HSCT.
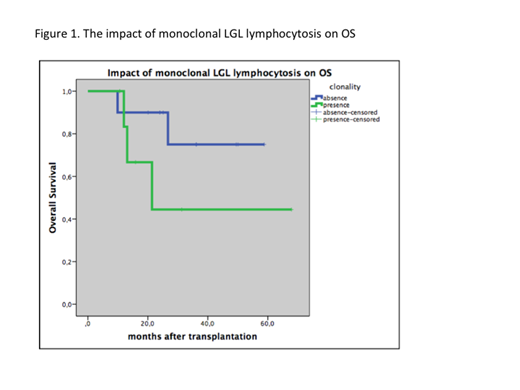
The median onset of LGL lymphocytosis was 11.5 (2.1- 55.7) months and median lymphocyte count at the time of diagnosis of LGL lymphocytosis was 5400/ mL (5170- 8700/ mL). None of the patient showed cytopenia, palpable splenomegaly, and none of them had typical signs or symptoms of an autoimmune disease. In addition; GvHD, viral infections, disease relapse and loss of donor chimerism were excluded during lymphocytosis. Samples from 19 patients were phenotyped by flow cytometry. These studies confirmed a T cell phenotype of LGLs in the majority of patients (n=12). Two patients presented with LGLs consistent with NK cells and seven showed properties of a mixed NK/T-cell lineage. A monoclonal LGL population of T-cell origin was identified in eight (42.1%) of these patients. With a median follow-up of 12.2 months none of the patients demonstrating increased LGL values has progressed to LGL leukemia or any other lymphoproliferative disorder. Four patients experienced cutaneous acute GVHD followed by a progressive chronic GVHD. Two patient developed a grade II acute cutaneous GVHD which rapidly responded to steroids in addition to cyclosporin A. Five patients had de novo chronic GVHD. In subgroup analysis, we compared the OS of monoclonal and oligoclonal LGL lymphocytosis and 1-year-OS was longer but non-significantly in monoclonal LGL lymphocytosis group; 75% ± 1.6% vs. 44.4% ± 2.2%, respectively (p= 0.21) (Figure). Median PFS was 28.8 months in oligoclonal LGL lymphocytosis group and 8.3 months in monoclonal LGL lymphocytosis group but the number of patients in this group does not provide enough statistical power to confirm whether the differences in PFS were statistically significant (p= 0.3). At the time of this report, three patients have died. The primary cause of death was relapse of the primary disease in one of the patients, whereas 2 patients died of TRM (10.5%).
Discussion: In conclusion, we observed LGL lymphocytosis in 2.8 % of a large cohort of post allo-HSCT survivors. Our data indicate that, even if monoclonal, post-transplantation LGL expansion may be considered as an expression of chronic stimulation triggered by allo-HSCT rather than the result of a malignant transformation.
Özcan:Amgen: Honoraria, Other: Travel support; BMS: Other: Travel support; Jazz: Other: Travel support; Sanofi: Other: Travel support; Bayer: Research Funding; Novartis: Research Funding; Roche: Other: Travel support, Research Funding; Archigen: Research Funding; Takeda: Honoraria, Other: Travel support, Research Funding; Abdi Ibrahim: Other: Travel support; MSD: Research Funding; AbbVie: Other: Travel support, Research Funding; Janssen: Other: Travel support, Research Funding; Celgene Corporation: Research Funding, Travel support. Ilhan:Roche: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; BMS: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; Janssen: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; Alexion: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau. Beksac:Amgen: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; Janssen: Research Funding, Speakers Bureau; Celgene: Speakers Bureau; Takeda: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal