

Background
Treatment of Multiple Myeloma (MM) has reached a point at which cure for a subset of patients becomes feasible. Still, resistance to therapy is a major challenge in the management of MM patients. Underlying resistance are the emergence of subclones, different to those found at diagnosis. Assessing the mutation status of recurrently mutated genes in MM can assist in tracking those subclones. In this study, we aimed to estimate the clonal composition by performing next generation sequencing in a set of samples from newly diagnosed patients included in a transplant-eligible trial (EMN02, Cavo et al., Blood 2017 130:397), and repeating this analysis in paired samples obtained at relapse when these patients were treated with Carfilzomib/ Pomalidomide/ Dexamethasone in the EMN11 trial (Sonneveld et al., Blood 2018 132:801).
Materials and methods
Panel-Seq was performed for a subset of most frequently recurring genes in MM (NRAS, KRAS, BRAF, DIS3, FAM46C and TP53). For 21 patients, DNA from bone marrow derived MM cells was available at diagnosis (EMN02/HOVON-95) and at relapse (EMN11/HOVON-114). For an additional 24 patients material at relapse was available (EMN11); survival data were available of 38/45 EMN11 cases. Endpoints were response, progression-free survival (PFS) and overall survival (OS). Response categories were stringent complete response (sCR), complete response (CR), very good partial response (VGPR), partial response (PR) and stable disease (SD). All samples had a sample purity > 53% (median 98% (53-100%)).
Results
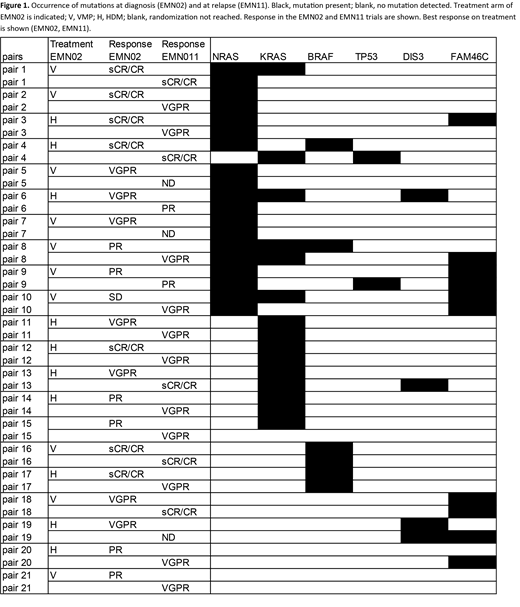
Using an allelic fraction of more than 1%, NRAS mutations were found in 48% of diagnostic samples, and in 43% of relapse samples (Figure 1). Overall, KRAS mutations were less frequent at relapse compared to diagnosis (24% vs 43% at diagnosis). In contrast, mutations in TP53 were found more frequently at relapse compared to diagnosis (in 2 patients at relapse (10%), compared to none at diagnosis). BRAF mutations were found in 4 patients at diagnosis and 2 at relapse (19% vs 10%). An allelic fraction of 40% or higher may be used to indicate clonality, i.e. all cells in the tumor are affected. For NRAS, this was observed at diagnosis and at relapse, in 5/10 and 5/9, and for KRAS, in 4/9 and 3/6, respectively.
In 9 out of 21 patients with paired samples identical NRAS mutations were detected at both time points. In 4 of these cases the estimated clone size of the NRAS mutation had increased at the time of relapse, while in 5 cases the clone remained similar in size. Four out of 6 cases with a stable clone size or diminishing NRAS clone had achieved sCR/CR with first-line treatment whereas all cases with increasing NRAS clone size were VGPR or worse (p=0.08). Interestingly, out of 19 cases with known survival data in the EMN11 trial, 2 out of 3 cases with an increase of 40% or more in NRAS clone size demonstrated an OS of less than 6 months, compared to only 1 out of 16 remaining cases. Four cases demonstrated both NRAS and KRAS mutations at diagnosis. In three of these cases, the KRAS mutation was not detected at relapse. In total, 5 pairs showed KRAS mutations at diagnosis and relapse. BRAF mutations were found in 4 cases at diagnosis; in two of these, both V600E, the mutation was also detected at relapse. None of the clonal shifts in genes other than NRAS demonstrated an association with PFS, OS or response.
Of the 45 patients analysed at entry in the EMN11 trial with an variant allelic frequency of 1% or higher, KRAS mutations were found in 36% of cases, followed by NRAS (29%), FAM46C (16%), BRAF (11%), TP53 (7%) and DIS3 (4%). Clonal NRAS mutations at baseline EMN11 cases were associated with worse OS but not PFS (median OS of clonal NRAS patients: 5 months compared to not reached for remaining patients; log rank p<0.05).
Conclusion
Although limited in patient size and number of genes studied, this unique cohort of homogeneously treated patients through 1st ánd 2nd line treatment allowed clinical analysis of mutations detected with high sensitivity to a variant allelic frequency of 1%. Sequential sequencing analysis has limited predictive value for 2nd line response, PFS and OS. Only an increase in NRAS clone size may be associated with worse survival, which requires validation in different datasets. In RRMM, clonal NRAS mutations may represent an additional risk factor.
Broijl:Takeda: Honoraria; Amgen: Honoraria; Janssen: Honoraria; Celgene: Honoraria. Zweegman:Celgene: Membership on an entity's Board of Directors or advisory committees, Research Funding; Takeda: Membership on an entity's Board of Directors or advisory committees, Research Funding; Janssen: Membership on an entity's Board of Directors or advisory committees, Research Funding. Minnema:Celgene Corporation: Honoraria, Research Funding; Amgen: Honoraria; Servier: Honoraria; Gilead: Honoraria; Jansen Cilag: Honoraria. Boccadoro:Celgene: Honoraria, Research Funding; Amgen: Honoraria, Research Funding; Janssen: Honoraria, Research Funding; Novartis: Honoraria, Research Funding; Bristol-Myers Squibb: Honoraria, Research Funding; AbbVie: Honoraria; Mundipharma: Research Funding; Sanofi: Honoraria, Research Funding. Sonneveld:Amgen: Honoraria, Research Funding; BMS: Honoraria; Celgene: Honoraria, Research Funding; Janssen: Honoraria, Research Funding; Karyopharm: Honoraria, Research Funding; Takeda: Honoraria, Research Funding; SkylineDx: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal