

Objective: Multiple myeloma (MM) is the second most common blood cancer in the United States. Clinical and pathological indicators are inadequate for accurate assessing prognosis of MM patients1. The objective of this study was to develop a robust computational model for predicting prognosis of MM patients by inferring transcription factor (TF) activities based on global gene expression measurements.
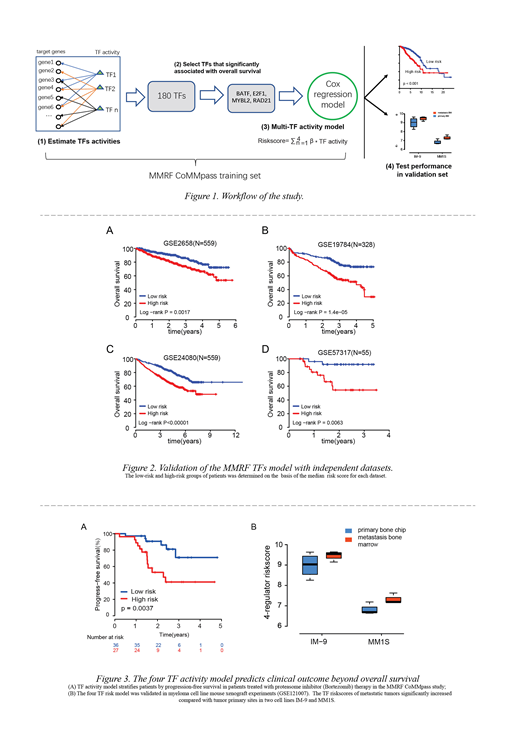
Methods: The workflow is illustrated in Figure 1. Gene expression data from MM patients were retrieved from the Multiple Myeloma Research Foundation CoMMpass study2 (MMRF) and Gene Expression Omnibus (GEO). MM-specific TF-target gene relationships were derived from the ENCODE database3 and co-expression analysis was based on MMRF data. The activity for each TF was inferred using a rank-based enrichment approach based on the target gene expression measurements in individual patients4. A prognostic model was developed using Cox regression analysis based on the inferred TF activities that demonstrated the highest association with patient survival. The TF model performance was further evaluated using data from four independent MM cohorts, in which gene expression levels were measured using different platforms.
Results: Based on MMRF data (767 patients), the inferred activity of four TFs, BATF, E2F1, MYBL2, and RAD21, showed the strongest association with patient survival. The derived prognostic model was validated in four independent MM data sets with a total of 1501 patients. Importantly, gene expression levels in the validation data sets were measured using microarray, which is different from the RNAseq used in training data sets, and supports the robustness of the TF activity model across different platforms (Figure 2). In addition, the TF activity model was able to predict clinical outcomes of patients that received bortezomib treatment, and it also revealed an association with metastasis in myeloma cell line xenograft experiments5 (Figure 3).
Conclusion: We developed a highly robust prognostic model for predicting MM clinical outcomes. The inferred TF activity further improves our understanding of the underlying regulatory basis of MM progression.
Reference
1. Rajkumar SV. Multiple myeloma: 2012 update on diagnosis, risk-stratification, and management. Am J Hematol. 2012;87(1):78-88.
2. l, a longitudinal study in multiple myeloma relating clinical outcomes to genomic and immunophenotypic profiles: Am Soc Hematology; 2013.
3. Consortium EP. The ENCODE (ENCyclopedia of DNA elements) project. Science 2004;306(5696):636-640.
4. Moerman T, Aibar Santos S, Bravo González-Blas C, et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 2018;35(12):2159-2161.
5. Shen Y, Mishima Y, Shi J, et al. Deciphering Clonal Evolution and Dissemination of Multiple Myeloma Cells In Vivo: Am Soc Hematology; 2018.
Abonour:Celgene: Consultancy, Research Funding; BMS: Consultancy; Takeda: Consultancy, Research Funding; Janssen: Consultancy, Research Funding. Roodman:Amgen: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal