

ZBTB7A is a transcription factor with function in hematopoietic lineage fate decisions (reviewed in Lunardi et al., 2013, Blood). Moreover, ZBTB7A has been shown to also be involved in regulation of glycolysis in solid tumors (Liu et al., 2014, Genes Dev.). Recently, we found ZBTB7A frequently mutated in acute myeloid leukemia (AML) with t(8;21) translocation. What is more, high expression of ZBTB7A correlated with better clinical outcome in cytogenetically normal AML (Hartmann et al., 2016, Nat Commun). The functional role of ZBTB7A and its alterations in myeloid malignancies, however, remains unclear, especially concerning the regulation of leukemia metabolism.
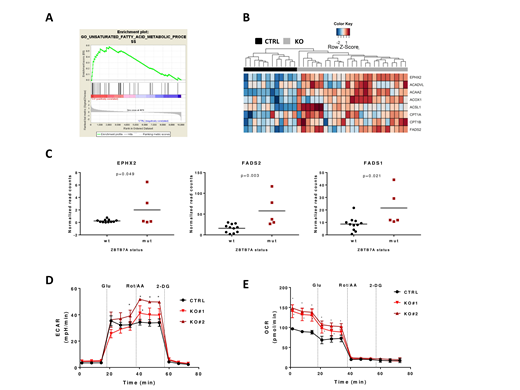
To investigate the effect of ZBTB7A mutations in leukemia, we generated K562 ZBTB7A knockout (KO) cells using CRISPR/Cas9, followed by RNA-Seq in KO and control cells. Thereby, we confirmed de-repression of the previously reported ZBTB7A target gene SLC2A3 (glucose transporter 3) as well as upregulation of several other glycolysis related genes (PGM2, PGM3, SLC2A1 and ENO2). ZBTB7A binding to all of these candidate target genes was validated using publicly available ChIP-Seq data from K562 cells (ENCSR000BME). Interestingly, Gene Set Enrichment Analysis revealed deregulation of distinct pathways related to fatty acid metabolism in this model (Figure 1A). KO cells overexpress genes involved in the fatty acid beta oxidation pathway (ACAA2, ACOX1, ACSL1, ACADVL, CPT1A and CPT1B) as well as genes related to other fatty acid metabolism (EPHX2, FADS2 and others) (Figure 1B). We could further validate ACAA2, ACOX1, ACADVL and CPT1A as direct ZBTB7A targets using ChIP-Seq data. Of special interest are CPT1A and CPT1B, which are targetable through Etomoxir treatment. KO cells showed an increased sensitivity to this drug compared to control (IC50= 120.6 and 125.5 µM in KOs vs 228.4 µM in control, p<0.0001). Moreover, analysis of RNA-Seq data from patients with AML t(8;21) revealed a significantly higher expression of EPHX2 (p=0.049), FADS2 (p=0.003) and FASD1 (p=0.021) in patients harboring ZBTB7A mutations (Figure 1C) using a two-tailed unpaired Student's t-test.
In order to evaluate our findings on a functional level, we performed metabolic flux assays in K562 ZBTB7A KO vs control cells. Using Seahorse technology (Agilent), we found that KO cells show a modest increase in extracellular acidification rate (ECAR), indicating a higher glycolysis. This effect becomes more obvious after mitochondrial respiratory chain inhibition: 41.20 and 51.66 mpH/min in KO clones vs 34.33 mpH/min in control (p=0.002 and p<0.001, respectively) (Figure 1D). This result suggests that loss of ZBTB7A may confer an advantage to cells in specific microenvironment with low oxygen availability, such as the bone marrow. Moreover, metabolic flux assays also revealed a nearly 50% increase in oxygen consumption rate (OCR) in KO cells after 1h glucose starvation: 140.53 and 148.53 pmol/min in KO clones vs 96.46 pmol/min in control (p<0.001 in both comparisons) (Figure 1E). Since the cells were deprived from glucose, the observed oxygen consumption may arise mainly from glutamate metabolism or fatty acid oxidation. Deprivation from glutamate reduced overall OCR but KO cells still showed increased oxygen consumption compared to control. These results therefore suggest that an increased beta oxidation of fatty acids leads to the higher OCR observed in KO cells.
In summary, we have demonstrated that the previously described role of ZBTB7A as a regulator of glycolysis in solid tumors is also relevant in myeloid malignancies. In addition, we identified the beta oxidative pathway and fatty acid synthesis as novel mechanisms underlying the perturbed function of ZBTB7A in tumor metabolism. ZBTB7A downregulation or mutation may lead to an increased energy production providing an advantage to leukemia cells. These findings likely have therapeutic implications, as metabolic inhibitors such as 2-deoxy-d-glucose and Etomoxir may specifically target ZBTB7A deficient malignancies.
Hiddemann:Celgene: Consultancy, Honoraria; Roche: Consultancy, Honoraria, Research Funding; Janssen: Consultancy, Honoraria, Research Funding; Gilead: Consultancy, Honoraria; Bayer: Research Funding; Vector Therapeutics: Consultancy, Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal