

Background. Relapse affects about 50% of AML patients who achieved remission after treatment, and the prognosis of relapsed AML is poor. Current evidence has shown that in many patients, mutations giving rise to relapse are already present at diagnosis and remain in small numbers in remission, defined as the minimal residual disease (MRD) [1]. Chemoresistant clones contributing to relapse of the disease arise from minimal residual disease (MRD) rather than resulting from newly acquired mutations during or after chemotherapy. MRD is the presence of measurable leukemic cells using non-morphologic assays. It is considered a strong predictor of relapse. The dynamics of clones comprising MRD is poorly understood and is considered influenced by a form of Darwinian selection.Methods. We propose a stochastic model based on a multitype (multi-clone) age-dependent Markov branching process to study how random events in MRD contribute to the heterogeneity in response to treatment in a cohort of six patients from The Cancer Genome Atlas database with whole genome sequencing data at two time points. Because human bone marrow cell counts are too large for direct stochastic simulation methods to be effective, we developed a hybrid numerical algorithm combining stochastic Gillespie-type and tau-leaping algorithms, and a deterministic differential equation solver, which uses much less computer time than a "straight Gillespie algorithm".
Results. We developed a stochastic model of clonal evolution based on a multitype (multi-clone) age-dependent Markov branching process model of cell proliferation. In brief, we consider the critical time interval between diagnosis and initial relapse of AML that includes cytotoxic chemotherapy, chemotherapy-induced myelosuppression and decrease in leukemic cells, non-leukemic marrow recovery, and growth of the leukemic clones due to refractory or relapsed disease. Underlying our model are assumptions regarding the structure of growth, differentiation, and competition of the normal and leukemic clones. Our model reflects the stochasticity inherent when leukemic clones are near depletion after chemotherapy, which we hypothesize strongly contributes to the interpatient heterogeneity in treatment response. The parameters are estimated by fitting the expected-value model to the patient's clinical data. The available data at diagnosis includes patient's weight, percent cellularity, white blood cell count, percentage of blasts in both peripheral blood and bone marrow, and percentage of normal neutrophils in the peripheral blood. Importantly, the time to relapse and percentage of blasts in bone marrow at relapse are available. The parameters fitted to the expected-value model offer an explanation of how a leukemic clone can escape chemotherapy and promote relapse. These clones have either high proliferation rates or high self-renewal rates. As a result, there is a range of different parameter combinations that can explain their ability to succeed. On the other hand, we also study the clones that have been eradicated by the time of relapse and conclude that these clones might be eliminated either because they are not competitive and therefore surrender to other clones, or they are simply killed by chemotherapy. Also, we checked if the parameters are biologically relevant by using the model to compute the corresponding clonal growth rates for each patient. That these values fit in the clinically observed range independently found in for patients with the NPM1 mutations [1], suggests that the model is consistent with clinical data.
Conclusions. Our model offers a more accurate understanding of how relapse arises and which properties allow a leukemic clone to thrive in the Darwinian competition among leukemic and normal hematopoietic clones. The model suggests a quantitative relationship between MRD and time to relapse and therefore may aid clinicians in determining when and how to implement treatment changes to postpone or prevent the time to relapse.
[1]Assessment of Minimal Residual Disease in Standard-Risk AML. N Engl J Med. 2016;374:422.
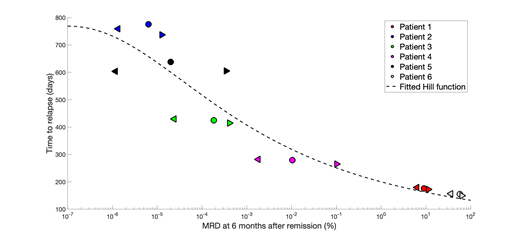
Relationship between MRD and time to relapse for all patients. Estimates of MRD and time to relapse are mean values from 1000 stochastic simulations. Each color corresponds to a single patient; left triangles, circles, and right triangles correspond to three parameter sets. Simulated points are fitted with a sigmoidal Hill function.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal