

Inherited disorders of fibrinogen are rare and can be quantitative (hypofibrinogenemia), qualitative (dysfibrinogenemia), or both (hypodysfibrinogenemia). Moreover, variants in fibrinogen have been described that associated with a bleeding, thrombotic, or combined phenotype. Although several genetic variations leading to hypodysfibrinogenemia have been identified, few clinically described cases have been molecularly characterized.
In this report we describe a 16-year-old female who presented with an unprovoked portal vein thrombosis. At the time of presentation, she appeared to have hypofibrinogenemia based on quantitation using the Clauss method. Immunoreactive testing demonstrated in a discrepancy between fibrinogen antigen and activity level consistent with dysfibrinogenemia. Single gene sequencing of FGA, FGB, and FGG revealed a heterozygous mutation in FGB, the gene encoding the fibrinogen beta chain (p.Tyr356Cys;). This variant (fibrinogen Villeurbanne) was previously described in a family with a clinical diagnosis of hypofibrinogenemia and a bleeding phenotype. This variant results in an unpaired cysteine residue near the beta chain hole, potentially resulting in abnormal protein synthesis or folding leading to intracellular protein degradation, protein instability within circulation, and/or abnormal polymer formation. It is also conceivable that this mutation could lead to aggregate formation via this unpaired cysteine between individual fibrinogen molecules or between fibrinogen and other proteins.
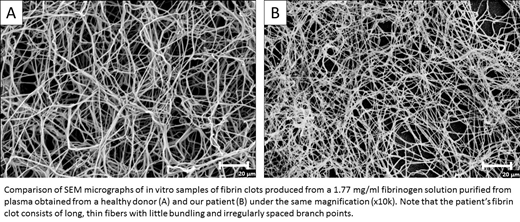
To better characterize this fibrinogen, we generated fibrin clots using purified fibrinogen from the patient and normal control. Clot structures were analyzed using scanning electron microscopy. The patient's fibrin clots, as compared to the control, were poorly formed, consisting of long, thin fibers with little bundling and irregular spaced branch points. These results indicate that the mutant fibrinogen is sufficiently stable in circulation to significantly impair polymer formation.
Albumin is the most prevalent plasma protein and is known to have an unpaired cysteine residue, making it a reasonable candidate to aggregate with the mutant fibrinogen. Plasma from the patient and control samples underwent co-immunoprecipitation followed by Western blot analyses in order to determine if we could detect albumin aggregation with the mutant fibrinogen. We found evidence of interaction between the mutant fibrinogen and albumin that was not observed in normal controls, suggesting that this patient's fibrinogen forms aggregates with albumin.
These findings contradict a previous conclusion that fibrinogen Villeurbanne results in hypofibrinogenemia, and indicate that it is a hypodysfibrinogenemia. Moreover, our laboratory findings together with the patient's presentation of an unprovoked portal vein thrombosis suggest the possibility that this mutant fibrinogen may, in some contexts, lead to pathological thrombosis.
Palumbo:Ionis Pharmaceuticals: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal