

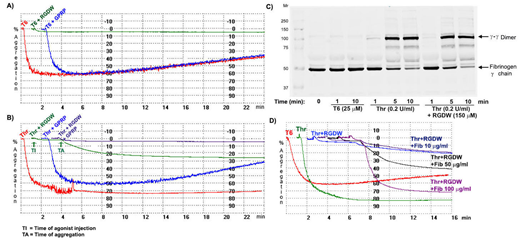
The formation of a stable platelet thrombus in vivo is accompanied by the generation of thrombin and the conversion of fibrinogen to cross-linked fibrin. This requires thrombin's action on fibrinogen, producing fibrin monomers, and factor XIII, catalyzing the reciprocal transamidation of the C-terminal γ-chain peptides from adjacent fibrinogen molecules. While the interaction of platelets with fibrinogen has been shown to be mediated by the interaction of fibrinogen γ-chain residues 404-411 with the RGD pocket on integrin αIIbβ3, the interaction with cross-linked fibrin is less well understood despite its clinical relevance. We previously reported that activated, but not unactivated, αIIbβ3 can support binding to D-dimer, the cross-linked plasmin fragment of fibrin, and 'D98' a fibrinogen fragment lacking the γ404-411 sequence. Here we studied the interaction of washed platelets in buffer containing 1 mM Mg2+ with polymerizing fibrin in an aggregometer by adding 0.2 U/ml thrombin and monitoring light transmission. To prevent the interaction of γ12 with the RGD pocket, studies were performed in the presence of 150 μM RGDW, a concentration that completely eliminates aggregation of the platelets to a thrombin receptor activating peptide (T6). RGDW eliminated the initial wave of platelet aggregation induced by thrombin, but did not prevent the appearance of a delayed wave (DW), which appeared after ~4-5 min (Panel A-B). Parallel immunoblot studies demonstrated that cross-linking of fibrinogen γ-chains, (γ-γ dimers) appeared within 1 min after adding thrombin and was ~85% complete by 5 min (Panel C). Thus, both cross-linked and uncross-linked fibrin(ogen) was present during the DW. Adding fibrinogen at 10-100 μg/ml enhanced the amplitude of the DW in a dose-dependent manner (Panel D). The fibrin polymerization inhibitor Gly-Pro-Arg-Pro (GPRP; 5 mM) dramatically inhibited the amplitude of the DW, supporting the importance of fibrin polymerization (Panel B). An antibody to platelet receptor α2β1 (6F1) had no effect on the slope or amplitude of the DW, whereas an antibody to GPIb (6D1) had a mild effect on the slope but not the amplitude of the DW. Since GPVI has variably been reported to interact with fibrin, we tested whether there was evidence of GPVI-mediated signaling during the DW. The known GPVI ligands/activators collagen and collagen-related peptide (CRP) (controls), produced marked increases 3 min after the onset of aggregation in phosphorylation of PLC-γ2-Y759, Syk-Y525/526, and LAT-Y191. Thrombin alone caused a major increase in phosphorylation of Syk-Y525/526 and a slight increase in phosphorylation of PLCγ2-Y759; however, adding RGDW dramatically inhibited the phosphorylation of Syk. We conclude that at 3 min after the onset of the DW there is minimal or no GPVI-mediated signal transduction. Similarly, neither the Src inhibitor PP2 at 100 μM or the Syk inhibitor VI at 1 μM, both of which inhibit CRP-induced platelet aggregation, inhibited the DW. Since polymerizing fibrin has the potential for multiple interactions with different platelet αIIbβ3 receptors, thus increasing the avidity, we compared the ability of αIIbβ3 antagonists to inhibit T6-induced aggregation versus the DW of polymerizing fibrin. Whereas the γ400-411 peptide was able to nearly completely inhibit T6-induced platelet aggregation at ~150 μM, it had virtually no impact on the DW at concentrations up to 500 μM. RGDW eliminated T6-induced aggregation at ~5 μM, but required 100-fold as much to produce ~80% inhibition of the DW. Similarly, eptifibatide was able to nearly eliminate T6-induced aggregation at ~5 μM, but 100-fold as much was required to eliminate the DW. Thus, even if αIIbβ3 antagonists do not inhibit the DW at doses that eliminate T6-induced aggregation, one cannot infer that other receptors are involved. Consistent with a dominant role for activated αIIbβ3 in the process, 10 mM EDTA essentially eliminated the DW and reptilase-induced fibrin polymerization did not lead to platelet incorporation unless the platelets were activated with T6 or another αIIbβ3 activator. In addition, both D-dimer and D98 inhibited the thrombin-induced DW at 200 µg/ml. We conclude that activated αIIbβ3 plays a dominant role in supporting high avidity platelet interactions with polymerizing, partially cross-linked, fibrin, and that the mechanism has features akin to, but distinct from the interaction of αIIbβ3 with fibrinogen.
Coller:CeleCor: Consultancy, Equity Ownership, Research Funding; Scholar Rock: Consultancy, Equity Ownership; Centocor/Janssen: Patents & Royalties: abxicimab; Accumetrics/Instrumentation Laboratory: Patents & Royalties: VerifyNow assay.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal