

In this issue of Blood, identify enhanced signaling through insulin-like growth factor 1 receptor (IGF1R) as the resistance mechanism to a phosphatidylinositol 3-kinase delta (PI3K-δ) inhibitor in a chronic lymphocytic leukemia (CLL) animal model. They also show that the inhibition of IGF1R provides a salvage treatment for PI3K-δ inhibitor–resistant tumors.1
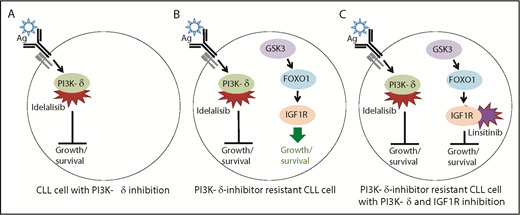
Model for the effects of the PI3K-δ inhibitor idelalisib and the IGF1R inhibitor linsitinib on PI3K-δ inhibitor–sensitive and –resistant CLL cells. (A) PI3K-δ is activated by the B-cell receptor (Ag, antigen) and inhibited by idelalisib, thus ablating the PI3K pathway-mediated growth and survival program that activates AKT signaling, which is required for CLL cell survival. (B) Upregulation of IGF1R expression via GSK3 and FOXO1 activates the MAPK pathway-mediated growth and survival program, thus leading to CLL cell survival. It is not known what leads to the activation of GSK3 and FOXO1 in PI3K-δ inhibitor–resistant CLL cells; however, FOXO1 activation is normally inhibited by AKT signaling downstream of the PI3K pathway. (C) Inhibition of IGF1R by linsitinib abolishes the MAPK-mediated growth and survival program, causing cell death in PI3K-δ–resistant CLL cells. The simultaneous inhibition of PI3K-δ is required because reactivation of PI3K/AKT signaling leads to FOXO1 inhibition and failure to upregulate IGF1R. In the serial-adoptive transfer and treatment experiments, Scheffold et al used the specific PI3K-δ inhibitor GS-649443 (rather than idelalisib) because this has favorable pharmacokinetic properties in mice.
Model for the effects of the PI3K-δ inhibitor idelalisib and the IGF1R inhibitor linsitinib on PI3K-δ inhibitor–sensitive and –resistant CLL cells. (A) PI3K-δ is activated by the B-cell receptor (Ag, antigen) and inhibited by idelalisib, thus ablating the PI3K pathway-mediated growth and survival program that activates AKT signaling, which is required for CLL cell survival. (B) Upregulation of IGF1R expression via GSK3 and FOXO1 activates the MAPK pathway-mediated growth and survival program, thus leading to CLL cell survival. It is not known what leads to the activation of GSK3 and FOXO1 in PI3K-δ inhibitor–resistant CLL cells; however, FOXO1 activation is normally inhibited by AKT signaling downstream of the PI3K pathway. (C) Inhibition of IGF1R by linsitinib abolishes the MAPK-mediated growth and survival program, causing cell death in PI3K-δ–resistant CLL cells. The simultaneous inhibition of PI3K-δ is required because reactivation of PI3K/AKT signaling leads to FOXO1 inhibition and failure to upregulate IGF1R. In the serial-adoptive transfer and treatment experiments, Scheffold et al used the specific PI3K-δ inhibitor GS-649443 (rather than idelalisib) because this has favorable pharmacokinetic properties in mice.
Cancer treatment with targeted therapies is undermined by the development of resistance to treatment in a fraction of patients. Thus, unraveling the resistance mechanisms is critical for the development of salvage therapies. Targeting of pathways comprising PI3K signaling has revolutionized the treatment of CLL and provides a paradigm of precision therapy2-4 for both the mode of action of pathway-specific drugs and for the identification of resistance mechanisms. Drugs inhibiting 2 components of the PI3K pathway, ibrutinib (targeting Bruton tyrosine kinase; BTK) and idelalisib (targeting PI3K-δ) are highly efficacious (see figure panel A). Previous studies have uncovered causes of ibrutinib resistance,5,6 but the mechanism of resistance to idelalisib has remained elusive.
Scheffold et al addressed this deficiency by modeling resistance to PI3K-δ inhibitors in vivo in 2 different murine serial-adoptive transfer and treatment models. Tumor cells derived from the Eμ-TCL1 CLL model were transplanted into syngeneic mice, and the Eμ-TCL1 mouse-derived CLL cell line TCL1-1927 was transplanted into NOD/SCID mice. The authors observed in both models that mice with CLL treated with a PI3K-δ inhibitor eventually succumbed to the disease despite an initial response. They then used the experimentally more amenable TCL1-192 line to develop a serial-transfer and treatment scheme to continuously treat CLL in vivo, thus generating tumors resistant to PI3K-δ inhibition that were then subjected to whole-exome sequencing (WES).
Naturally, the expectation was to identify mutations in PI3K-δ that abolished the action of the inhibitor or recurrent genetic alterations in pathway genes downstream of PI3K-δ. However, the WES analysis did not identify any single recurrent mutation in PI3K-δ, which was particularly surprising because mutations in BTK at the inhibitor-binding site are the hallmark of ibrutinib-resistant CLL.5,6 In addition, no recurrent mutations were found in other genes. Similarly, CLL cells from resistant patients were found to be devoid of unifying recurrent mutations that could explain drug resistance (Ghia et al8 ; Paolo Ghia, San Raffaele Hospital, Milan, Italy, personal communication, 14 April 2019).
Scheffold et al then transcriptionally profiled the tumors and identified several genes whose expression was deregulated in PI3K-δ inhibitor–resistant vs PI3K-δ inhibitor–sensitive tumors, with upregulated expression of IGF1R showing the strongest association with resistance. They then demonstrated that this upregulation indeed contributed to PI3K-δ inhibitor resistance (see figure panel B). But how is IGF1R upregulation in the resistant cells mediated?
Based on a publication showing that IGF1R can be transcriptionally activated by FOXO1 and GSK3,9 the authors investigated the potential role of these proteins in PI3K-δ inhibitor resistance. Upregulation of IGFIR expression was found to be attenuated by inhibition of FOXO1 or GSK3, indicating that enhanced IGF1R expression is at least partially mediated by GSK3 activity that directly or indirectly causes increased nuclear localization of FOXO1. Further downstream, IGF1R promotes CLL cell growth through activation of the MAPK pathway.
Next, Scheffold et al sought to investigate whether PI3K-δ inhibitor resistance can be overcome by inhibition of IGF1R with linsitinib. They demonstrated that treatment with the combination of linsitinib with PI3K-δ inhibitors, but not linsitinib-only treatment, was toxic for PI3K-δ inhibitor–resistant CLL cells (see figure panel C). Linsitinib-only treatment did not impair tumor cell growth because IGF1R expression was downregulated in PI3K-δ inhibitor–resistant cells in the absence of PI3K-δ inhibitor. This downregulation is likely a result of reactivation of PI3K/AKT signaling, which inhibits nuclear translocation of FOXO1. Conversely, with combined treatment, PI3K/AKT inhibition leads to GSK3 and FOXO1 activation that enhances expression of IGF1R protein, which is then targeted by linsitinib. Overall, the results indicate that IGF1R upregulation promotes cell growth of PI3K-δ inhibitor–resistant CLL cells via activation of the MAPK pathway and that PI3K-δ inhibitor resistance could be overcome by simultaneous PI3K-δ/IGF1R inhibition.
What is the translational aspect? Ideally, idelalisib-resistant CLL cells derived from patients would be analyzed. This could validate (or refute) the clinical relevance of the Scheffold et al findings. This undertaking is the authors’ future aim, and preliminary evidence indicates that they may be on the right track. The authors could demonstrate in a CLL sample from 1 patient with high IGF1R expression after 10 months of idelalisib treatment that those cells showed a low response to PI3K-δ inhibitor in vitro. In addition, treatment with a combination of idelalisib and linsitinib synergistically impaired cell growth, indicating that co-inhibition of IGF1R can sensitize tumor cells with reduced sensitivity to PI3K-δ inhibitors. Notably, among patients with untreated CLL, a sizable fraction showed high IGF1R expression levels, particularly in association with trisomy 12. Those patients may benefit from combination treatment with idelalisib and linsitinib.
The work of Scheffold et al highlights the importance of animal models in identifying drug-resistance mechanisms. Although it remains to be determined to what extent the newly identified IGF1R-mediated PI3K-δ inhibitor resistance mechanism plays a role in idelalisib resistance in human CLL, the results provide an indication of what to watch for in clinical trials. The study also emphasizes that resistance mechanisms may not be restricted solely to genetic mutations. One caveat for murine modeling of drug resistance is the much shorter time span of disease progression compared with that in humans and the impact this may have on clonal evolution of the tumor. This could be particularly relevant in the case of PI3K-δ inhibition by idelalisib (but not ibrutinib10 ), which has been reported to increase genomic instability in B cells and may thus introduce mutations by enhanced activity of activation-induced cytidine deaminase, which normally initiates immunoglobulin somatic hypermutation and class switching, in off-target genes.11 Because idelalisib can be administered over a long period of time, there is the potential to uncover additional resistance mechanisms that involve genetic alterations that activate tumor-promoting pathways. It will be interesting to integrate the findings of the genomic analyses by Scheffold et al in mice and Ghia et al8 in humans because this may uncover pathways that contribute to PI3K-δ inhibitor resistance in some patients with CLL. This well-conducted study has provided a wealth of information and intriguing new leads that will be instrumental in overcoming the problem of idelalisib resistance in human CLL and other malignancies in which PI3K-δ plays a pathogenic role. In the short term, the primary focus should be on determining whether linsitinib is the ideal companion for idelalisib in the treatment of lymphoid tumors in which IGF1R is upregulated because of PI3K-δ inhibitor resistance.
Conflict-of-interest disclosure: The author declares no competing financial interests.
