

In this issue of Blood, 1 describe novel pathways responsible for self- and cross-tolerance of monocytes evoked by endotoxin stimulation or by endogenous Toll-like receptor 4 (TLR4) ligands (see figure). This study helps unravel the pathways of innate immune memory of monocyte hyporesponsiveness (see figure), which occur in systemic inflammatory response syndrome (SIRS) and contribute to development of secondary infections and severe sepsis.
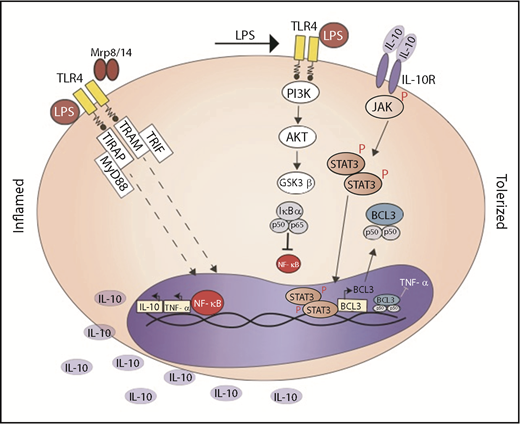
LPS or Mrp-8 activates TLR-4 and downstream NF-κB translocation into the nucleus; in the nucleus, NF-κB induces the synthesis of TNF-α and IL-10. Secondary stimulation with LPS activates PI3K/AKT and phosphorylates GSK3β; inactive phosphorylated GSK3β does not degrade IκBα and impairs NF-κB activation. IL-10, via IL-10R, phosphorylates STAT3 that induces BCL-3 synthesis. BCL-3 promotes binding of inactive NF-κB to the TNF promoter. R, receptor
LPS or Mrp-8 activates TLR-4 and downstream NF-κB translocation into the nucleus; in the nucleus, NF-κB induces the synthesis of TNF-α and IL-10. Secondary stimulation with LPS activates PI3K/AKT and phosphorylates GSK3β; inactive phosphorylated GSK3β does not degrade IκBα and impairs NF-κB activation. IL-10, via IL-10R, phosphorylates STAT3 that induces BCL-3 synthesis. BCL-3 promotes binding of inactive NF-κB to the TNF promoter. R, receptor
SIRS is characterized by an intense activation of the innate immune system, either from activation of pathogen-associated molecular patterns or damage-associated molecular patterns. The latter’s activation explains the immune stimulation in absence of pathogens, caused by the release of endogenous nuclear, mitochondrial, or cytosolic molecules, collectively named alarmins. Myeloid-related proteins-8 (Mrp-8) and Mrp-14 alarmins are released by necrotic cells or secreted by phagocytes when tissue damage occurs, form heterodimers (Mrp-8/14), and activate TLR4.
The profound activation of the innate system triggered by endotoxins or alarmins during SIRS is followed by the compensatory anti-inflammatory response syndrome, which helps to avoid extensive tissue damage from inflammation. Nevertheless, compensatory anti-inflammatory response syndrome contributes to the development of opportunistic secondary infection and sepsis, and occurs from inactivation of monocytes, rather than paralysis of other phagocytes. Hyporesponsive monocytes express HLA-DRlow, produce lower amounts of inflammatory mediators, higher amounts of anti- inflammatory mediators, and display impaired phagocytic activity. Different mechanistic pathways have been described for monocyte hyporesponsiveness, such as increased expression of negative pro-inflammatory TLR signaling pathways leading to impaired NF-κB activation and expression of anti-inflammatory heat-shock proteins.2 The incomplete blockade of self- or cross-monocyte tolerance by blocking known mechanisms suggests there are multiple and independent intracellular pathways involved with inactivation.
In the Freise et al study, elegant and robust in vitro and ex vivo experimental approaches in mice and human monocytes were carried out to define the intracellular pathways activated by lipopolysaccharide (LPS) or alarmin Mpr-8 in monocyte tolerance to LPS. Pharmacological and genetic manipulations in the used models confirmed the pathways. Translational data from humans with sterile SIRS evoked by cardiopulmonary bypass surgery also corroborate the findings.
LPS or Mrp-8 preactivation induced early and long-lasting inactivation of glycogen synthase kinase (GSK-3β), a process dependent on persistent activation of phosphatidylinositol 3-kinase (PI3K)/AKT pathway. Indeed, in vitro and ex vivo monocyte hyporesponsiveness was induced by pharmacological blockade of GSK-3β, and inhibiting AKT evoked complete blockade of the hyporesponsive state. Interestingly, MAPK p38 was activated in hyporesponsive monocytes, pointing out the specificity of the pathway PI3K/AKT/GSK-3β. Furthermore, increased intracellular levels of IκBα, a GSK-3β substrate, confirmed the inactivation of GSK-3β. Because IκBα downregulated NF-κB, the PI3K/AKT/GSK-3β/NFκB pathway was proposed as a potential mediator in monocyte hyporesponsiveness. The specificity of AKT/GSK-3β in the anti-inflammatory axis has been demonstrated in diverse models of phagocytes activation. Conversely, recent data showed self- and cross-hyporesponsiveness evoked by Mrp-8 prestimulation in human and both murine peritoneal or bone marrow derived macrophages is primarily through downregulation of phosphorylated p38 MAPK rather than the inactivation of NF-κB.3 Given the similarity of the experimental aims in both studies, the results suggest that different concentrations and schedules of stimulation and restimulation may activate distinct intracellular pathways.
It is noteworthy that in the Freise et al study, LPS or Mrp-8 tolerized monocytes expressed high levels of B-cell lymphoma-3 (BCL-3), also a member of the IκB family of NF-κB regulatory protein. Furthermore, clear involvement of BCL-3 in hyporesponsiveness was shown by normal response of BCL-3−/− Hoxb8 cells to a second stimulation of LPS. A possible connection between GSK-3β and BCL-3 was prevented because pharmacological blockade of GSK-3β did not cause overexpression of BCL-3. Meanwhile, gene expression of BCL- 3 was induced for 4 and 24 hours after monocyte stimulation by endotoxin or alarmin, and an additional transcriptional mechanism was then proposed for monocyte hyporesponsiveness. A connection between IL-10 and BCL-3 had already been suggested in models of immune tolerance, and Chuang et al4 demonstrated BCL-3 is a key protein in IL-10 induced immunosuppressed macrophages. Moreover, IL-10 secretion is enhanced by TLR-4 stimulation and higher systemic levels of the cytokine are found in SIRS models.5 Data from the Fraise study showed IL-10, via IL-10 receptor, caused downstream STAT-3 phosphorylation, which triggered BCL-3 expression in LPS or Mrp-8 tolerized monocytes. Early and long-lasting STAT-3 phosphorylation was detected in in vitro and in vivo tolerized monocytes. Pharmacological blockade of STAT-3 reduced messenger RNA BCL-3 levels and reversed LPS or Mrp-8 induced human monocytes hyporesponsiveness. Likewise, monocytes of patients with dominant-negative mutations in the STAT-3 gene were not self-tolerant to LPS. The translational confirmation of IL-10/STAT3/BCL-3 in monocyte hyporesponsiveness was demonstrated in a model of sterile SIRS induced by cardiopulmonary bypass surgery. Long-lasting, elevated serum levels of Mrp8/14 were detected after bypass completion in association with higher levels of IL-10. In the meantime, monocytes from patients had high levels of phosphorylated STAT-3 and were hyporesponsive to ex vivo stimulation with LPS. Although Fraise and coauthors present convincing data for the proposed pathway, a recent study did not corroborate STAT3 phosphorylation by IL-10 in ex vivo stimulated monocytes collected from SIRS and septic patients.6 It is important to take into account the diversity of alarmins released in the different damaged tissues, which are released by different mechanisms, and induce distinct pathways of cell activation. The proposed mechanisms in the Freise et al study occur in in vitro and in vivo TLR4 activation by LPS or Mrp-8 alarmins, and further investigation must be carried out to elucidate the participation of the pathways in monocyte tolerance evoked by other alarmins. Data will be pivotal to corroborate the proposal of PI3K/AKT/GSK-3β/NF-κB and IL-10/STAT3/BCL3 pathways as targets for reversal of monocyte tolerance.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal