

In a study published in this issue of Blood, describe high efficacy and safety of sutimlimab, a humanized monoclonal antibody to complement protein 1s (C1s), in the therapy of cold agglutinin disease (CAD).1 If confirmed, these findings represent a new breakthrough in the management of CAD, an autoimmune hemolytic anemia that is often difficult to treat.
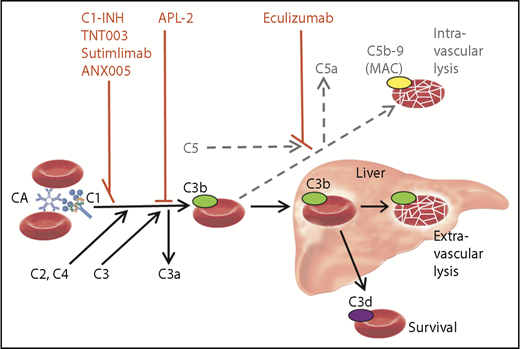
Role of the classical and terminal complement activation pathways in CAD and targets of complement inhibitors. Only relevant details are shown. Black arrow, major pathway; gray/dotted arrow, minor pathway; C1-INH, plasma-derived C1 esterase inhibitor; MAC, membrane attack complex.
Role of the classical and terminal complement activation pathways in CAD and targets of complement inhibitors. Only relevant details are shown. Black arrow, major pathway; gray/dotted arrow, minor pathway; C1-INH, plasma-derived C1 esterase inhibitor; MAC, membrane attack complex.
CAD, the well-defined clinicopathologic entity investigated by Jäger et al, should be distinguished from secondary cold agglutinin syndrome (CAS), a similar cold hemolytic syndrome occasionally complicating specific infections or aggressive lymphoma.2 The pathogenesis in CAD can be considered made up of 2 steps: (1) CAD is a distinct, B-cell lymphoproliferative bone marrow disorder in which the clonal cells produce a cold agglutinin (CA), usually an immunoglobulin M3 ; and (2) this results in an entirely complement-mediated, classical pathway-dependent autoimmune hemolysis.4
Drug therapy is not always indicated. Anemia can be severe, however, and the burden of symptoms has probably been underestimated. Retrospective studies have demonstrated the severity of the symptoms; pharmacological treatment has been given in 70% to 80% of the patients.5 Until recently, all effective therapies have been directed against the pathogenic B-cell clone. Rituximab monotherapy is the most accepted approach, although response rates and duration are limited, whereas rituximab plus bendamustine combination therapy has proved highly efficient, often yielding complete and durable responses.6
The first complement-directed therapeutic agent for CAD to be described in a prospective trial was the anti-C5 monoclonal antibody eculizumab, which was found to have a significant effect on intravascular hemolysis but only a modest effect on hemoglobin levels.7 The obvious explanation is that eculizumab will not target the predominant hemolytic pathway in CAD (see figure). It has been hypothesized, therefore, that upstream classical pathway inhibition would do better. According to a case report describing a patient with severe CAS, treatment with high doses of C1-INH promptly abrogated the hemolysis8 ; however, long-term therapy with this substance will probably not be feasible because patients with CAD have normal endogenous C1-INH levels.
A study published in 2014 found that TNT003, the murine precursor of sutimlimab, was able to efficiently inhibit C1 and prevent in vitro deposition of C3 fragments on erythrocytes in the presence of patient sera as a source of CA.4 The addition of TNT003 also resulted in abrogation of hemolysis and prevention of erythrophagocytosis by a phagocytic cell line.
In the clinical phase 1b trial by Jäger and colleagues, 10 patients with CAD received therapy with sutimlimab (previously termed TNT009 or BIVV009), starting with a test dose and followed by weekly doses of 60 mg/kg. The participants were vaccinated against meningococci, pneumococci, and Haemophilus but did not receive any pharmacological prophylaxis. The treatment was well tolerated without any convincing drug-related adverse events. Seven patients (70%) responded, as defined by a hemoglobin increase of ≥2 g/dL. The median increase in hemoglobin levels was 1.6 g/dL within the first week and 3.9 g/dL within 6 weeks. Bilirubin levels normalized within 24 hours in most patients. Relapses were observed 3 to 4 weeks after the last dose, but reexposure to sutimlimab restored the control of hemolytic anemia. All previously transfused patients became transfusion independent. Of the 3 nonresponders, at least 2 patients turned out not to have typical CAD (1 had a MYD88 L265P–positive lymphoplasmacytic lymphoma of the bone marrow and 1 was classified as having a mixed autoimmune hemolytic anemia). The authors conclude that upstream inhibition of the classical complement pathway is an effective treatment of CAD.
This high-efficacy and favorable safety profile will need to be confirmed by at least 1 larger trial. Two such phase 2 and 3 trials are currently being conducted (ClinicalTrials.gov, #NCT03347396 and #NCT03347422).
Sutimlimab is not the only novel complement inhibitor that has entered clinical trials. An in vitro study has shown prompt inhibition of classical pathway activation and CA-induced hemolysis in the presence of ANX005, a humanized monoclonal antibody to C1q.9 A safety study in healthy volunteers has been completed but still not published. APL-2 is a compstatin analog, a pegylated cyclic peptide that prevents the cleavage of C3 into C3a and C3b and is designed for subcutaneous administration. This substance has shown favorable results in a phase 1b trial in patients with paroxysmal nocturnal hemoglobinuria and has recently entered clinical trials in autoimmune hemolytic anemia, including CAD (ClinicalTrials.gov, #NCT03226678).10
Complement-directed therapy for CAD will encounter some limitations. First, in contrast to chemoimmunotherapy, treatment will probably have to continue infinitely to maintain its effect. Second, symptoms caused directly by the red blood cell agglutination, such as acrocyanosis and Raynaud-like phenomena, are not complement mediated and will not be relieved. On the other hand, the B-cell–directed therapies also have obvious limitations: at least 25% of the patients will not respond; the time to response is up to several months6 ; and some patients have contraindications or are reluctant to receive treatment with cytotoxic drugs. In contrast, sutimlimab is rapidly acting and seems to have a very favorable toxicity profile. It is beyond any doubt, therefore, that the upstream complement inhibitors have the potential to fill a substantial unmet need among patients with CAD. In severe cases and acute exacerbations, such therapy may, it is hoped, provide a “bridge” that will allow rapid achievement of remission and transition to B-cell–directed treatment when the situation is under control. Furthermore, the potential of proximal complement inhibition in secondary CAS and paroxysmal cold hemoglobinuria remains to be explored.
Conflict-of-interest disclosure: S.B. has received research support from Mundipharma, travel support from Alexion, lecture honoraria from Bioverativ and Janssen-Cilag, and has consulted for Apellis, Bioverativ, Momenta Pharmaceuticals, and True North Therapeutics.
