

TO THE EDITOR:
Survival estimates of adults with sickle cell disease (SCD) in high-income countries are limited because of the absence of prospective cohorts of young adolescents followed to and through adulthood. Recently, Gardner et al described a cohort of 712 adults with SCD (16-80 years of age) at King’s College Hospital (London, U.K.) from 2004 to 2013 that included 5268 patient-years of observation, and the median observation per patient was 8 years.1 The estimated median age of survival was 67 years for 450 adults with HbSS/HbSβ0, an age that is significantly greater than that observed in a similar study in the United States reporting a median survival of 44.7 years for 132 adults with HbSS/HbSβ0/HbSD.2
To address whether the higher median survival of adults with SCD living in London compared with adults with SCD living in the United States was related to statistical methods, and not cohort characteristics, we conducted a pooled analysis from 2 academic SCD centers in the United States. The median survival was determined for adults with SCD who received medical care at Vanderbilt University Medical Center (VUMC) and the University of North Carolina (UNC). Statistical analysis included summaries of demographic and clinical variables, with comparisons by cohort and phenotype, as well as Kaplan-Meier analysis to estimate median survival age with 95% confidence intervals (95% CIs), using entry age as the baseline, which adjusts for left truncation and is a conservative estimate for survival.3 As a less conservative statistical strategy to estimate median survival, similar to Gardner et al, we calculated the median survival from birth.1 Our primary focus was not to identify risk factors for mortality in adults with SCD, because these results have been reported,2,4 but to estimate the median survival, using a more conservative analytical strategy from data at 2 academic sites providing medical care for adults with SCD in the United States.
The study period was from September 2003 to January 2016 for VUMC4 and from August 2004 to May 2012 for UNC.2 The minimum age of inclusion into the analysis was 18 years, and the minimum duration of follow-up was 6 months. Eleven individuals were excluded from the analysis because they were followed for <6 months; among this group, 45.5% died (5/11) at a median age of 42.6 years (range, 20.0-79.4). The analysis included 300 individuals, accounting for 2017.5 patient-years of observation and a median observation of 6.8 years per patient.
Forty-six deaths occurred among 300 individuals at a median age of 44.6 years (interquartile range, 34.6-55.1). The median baseline age of the 2 cohorts was 25.1 and 35.4 years for VUMC and UNC, respectively. The median ages of survival for HbSS/HbSβ0/HbSD and HbSC/HbSβ+, based on the pooled Kaplan-Meier estimates and adjusted for entry age, were 48.0 years (95% CI, 44.4-58.4) and 54.7 years (95% CI, 38.6-62.9), respectively (Figure 1); the 2 distributions were not different (P = .889, log-rank test). If we included the 11 individuals originally excluded because they were followed for <6 months, the median survival for HbSS/HbSβ0/HbSD decreased by 1.7 years but remained unchanged for HbSC/HbSβ+. Conditional survival increased if participants lived to 18, 25, and 30 years of age, with an inverse relationship between age attained and subsequent median survival (Table 1). In contrast, using the statistical approach of Gardner et al,1 we obtained similar results for median survival from birth without adjustment for entry age; the estimate was 66.1 years (95% CI, 55.5-76.7) years for HbSS/HbSβ0/HbSD and 62.9 years (95% CI, 56.5-69.3) for HbSC/HbSβ+. The survival distributions were not different (P = .412, log-rank test).
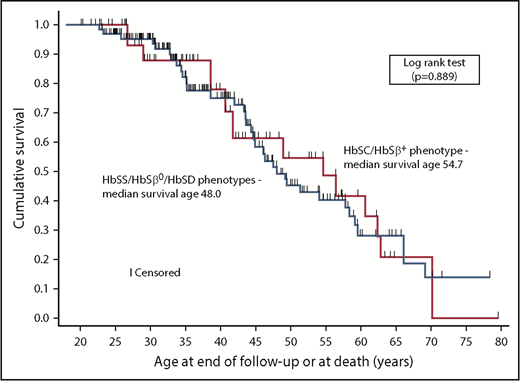
Kaplan-Meier curves depicting survival of adults with SCD. Median survival was 48.0 years (95% CI, 44.4-58.4) for individuals with the HbSS/HbSβ0/HbSD phenotypes (n = 225) and 54.7 years (95% CI, 38.6-62.9) for individuals with the HbSC/HbSβ+ phenotypes (n = 75).
Kaplan-Meier curves depicting survival of adults with SCD. Median survival was 48.0 years (95% CI, 44.4-58.4) for individuals with the HbSS/HbSβ0/HbSD phenotypes (n = 225) and 54.7 years (95% CI, 38.6-62.9) for individuals with the HbSC/HbSβ+ phenotypes (n = 75).
Characteristics of adults with SCD in the VUMC and UNC cohorts (N = 300)
| Patient characteristics | All patients (N = 300) | VUMC (n = 150) | UNC (n = 150) | P (cohorts)* | HbSC/HbSβ+ (n = 75) | HbSS/HbSβ0/HbSD (n = 225) | P (phenotypes)* |
|---|---|---|---|---|---|---|---|
| Age at baseline, median (IQR), y | 28.9 (22.5-40.6) | 25.1 (18.7-31.6) | 35.4 (26.0-43.8) | <.001† | 31.1 (23.7-43.8) | 28.6 (22.0-39.5) | .150† |
| Follow-up, mean (SD), y | 6.7 (3.3) | 7.1 (3.7) | 6.3 (2.9) | .045 | 6.6 (3.4) | 6.8 (3.3) | .779 |
| Males, % | 43.7 | 48.0 | 39.3 | .130 | 48.0 | 42.2 | .382 |
| SCD phenotype (HbSS/HbSβ0/HbSD), % | 75.0 | 68.7 | 81.3 | .011 | NA | NA | NA |
| Hemoglobin, mean (SD), g/dL | 9.5 (1.8) | 9.9 (1.8) | 9.2 (1.8) | .001 | 11.2 (1.6) | 9.0 (1.5) | <.001 |
| Reticulocyte, mean (SD), % (n = 265) | 8.0 (5.6) | 9.0 (6.5) | 7.2 (4.6) | .009 | 4.2 (3.0) | 9.2 (5.7) | <.001 |
| On hydroxyurea, % | 58.7 | 60.0 | 57.3 | .639 | 29.3 | 68.4 | <.001 |
| Death, % | 15.3 | 14.7 | 16.0 | .749 | 16.0 | 15.1 | .853 |
| Median age of survival for adults with SCD, by starting initial survival age (N = 300) | |||||||
| Median (95% CI): survival to ≥18 y | 49.2 (44.4-58.4) (n = 300) | 43.6 (40.6-59.5) (n = 150) | 54.1 (46.3-62.9) (n = 150) | .191‡ | 54.7 (38.6-62.9) | 48.0 (44.4-58.4) | .889‡ |
| Median (95% CI): survival to ≥25 y | 54.7 (46.3-60.6) (n = 198) | 54.7 (43.3-60.6) (n = 80) | 54.1 (46.3-62.9) (n = 118) | .858‡ | 56.5 (38.6-62.9) (n = 53) | 51.4 (44.9-59.5) (n = 145) | .926‡ |
| Median (95% CI): survival to ≥30 y | 56.5 (46.3-60.6) (n = 138) | 54.7 (40.6-undefined) (n = 42) | 56.5 (46.4-62.9) (n = 96) | .815‡ | 54.7 (38.6-62.9) (n = 38) | 54.1 (46.1-66.1) (n = 100) | .760‡ |
| Patient characteristics | All patients (N = 300) | VUMC (n = 150) | UNC (n = 150) | P (cohorts)* | HbSC/HbSβ+ (n = 75) | HbSS/HbSβ0/HbSD (n = 225) | P (phenotypes)* |
|---|---|---|---|---|---|---|---|
| Age at baseline, median (IQR), y | 28.9 (22.5-40.6) | 25.1 (18.7-31.6) | 35.4 (26.0-43.8) | <.001† | 31.1 (23.7-43.8) | 28.6 (22.0-39.5) | .150† |
| Follow-up, mean (SD), y | 6.7 (3.3) | 7.1 (3.7) | 6.3 (2.9) | .045 | 6.6 (3.4) | 6.8 (3.3) | .779 |
| Males, % | 43.7 | 48.0 | 39.3 | .130 | 48.0 | 42.2 | .382 |
| SCD phenotype (HbSS/HbSβ0/HbSD), % | 75.0 | 68.7 | 81.3 | .011 | NA | NA | NA |
| Hemoglobin, mean (SD), g/dL | 9.5 (1.8) | 9.9 (1.8) | 9.2 (1.8) | .001 | 11.2 (1.6) | 9.0 (1.5) | <.001 |
| Reticulocyte, mean (SD), % (n = 265) | 8.0 (5.6) | 9.0 (6.5) | 7.2 (4.6) | .009 | 4.2 (3.0) | 9.2 (5.7) | <.001 |
| On hydroxyurea, % | 58.7 | 60.0 | 57.3 | .639 | 29.3 | 68.4 | <.001 |
| Death, % | 15.3 | 14.7 | 16.0 | .749 | 16.0 | 15.1 | .853 |
| Median age of survival for adults with SCD, by starting initial survival age (N = 300) | |||||||
| Median (95% CI): survival to ≥18 y | 49.2 (44.4-58.4) (n = 300) | 43.6 (40.6-59.5) (n = 150) | 54.1 (46.3-62.9) (n = 150) | .191‡ | 54.7 (38.6-62.9) | 48.0 (44.4-58.4) | .889‡ |
| Median (95% CI): survival to ≥25 y | 54.7 (46.3-60.6) (n = 198) | 54.7 (43.3-60.6) (n = 80) | 54.1 (46.3-62.9) (n = 118) | .858‡ | 56.5 (38.6-62.9) (n = 53) | 51.4 (44.9-59.5) (n = 145) | .926‡ |
| Median (95% CI): survival to ≥30 y | 56.5 (46.3-60.6) (n = 138) | 54.7 (40.6-undefined) (n = 42) | 56.5 (46.4-62.9) (n = 96) | .815‡ | 54.7 (38.6-62.9) (n = 38) | 54.1 (46.1-66.1) (n = 100) | .760‡ |
IQR, interquartile range; NA, not applicable; SD, standard deviation.
χ2 test for categorical variables and Student t test for continuous variables, unless otherwise indicated.
Mann-Whitney U test.
Log-rank test for comparison of survival distribution.
A multivariable Cox regression analysis for survival, including entry age and adjusting for cohort (VUMC vs UNC), revealed that entry age was directly related to risk of death (hazard ratio [HR], 1.04; 95% CI, 1.02-1.07; P = .001); whereas, baseline hemoglobin level was inversely related to the risk of death (HR, 0.74; 95% CI, 0.60-0.92; P = .006). The following covariates were not associated with an increased HR for death: sex (HR = 1.26; 95% CI, 0.69-2.29; P = .454), SCD phenotype (HbSS/HbSβ0/HbSD) (HR, 0.66; 95% CI, 0.26-1.67; P = .385); and hydroxyurea therapy (HR, 0.96; 95% CI, 0.48-1.94; P = .920). Hemoglobin F levels were not uniformly collected across both cohorts.
As with any retrospective survival analysis in young adults with a life-threatening disease, left truncation bias likely occurred, and adjustment for entry age does not remove all bias. The better alternative is to create a statewide or national population-based registry in which individuals with SCD are followed from birth or from young adolescence, at which point the current mortality rates are <1%.5,6 However, short of a population-based registry, adjustment for left truncation provides a more conservative estimate of the median survival. This pooled analysis did not include extensive risk factors for death. Both published studies from VUMC and UNC had more complete evaluations for risk factors associated with death.2,4 In both cohorts, the proportion of participants with HbSS/HbSβ0/HbSD prescribed hydroxyurea was ∼60%, much higher than the 10% reported in the London cohort; however, the use of hydroxyurea in our pooled analysis was not associated with increased age of survival. We did not assess the dose of hydroxyurea or adherence to therapy. Both dose and adherence to hydroxyurea may have influenced the postulated impact of hydroxyurea therapy.
The primary outcome of our survival analysis included adjustment for left truncation, a conservative and, we believe, a more appropriate estimate of survival in adults with SCD.3 Future studies evaluating overall survival in adults with SCD should at least include adjustment for left truncation bias, allowing results to be compared across studies. At 2 academic medical centers with a comprehensive approach to the medical care of adults with SCD in the United States, the median survival for adults with SCD is ≥20 years shorter than for African Americans living in the United States.7
Acknowledgments
The authors thank members of the Vanderbilt-Meharry Sickle Cell Disease Center of Excellence and the DeBaun laboratory.
This work was funded by Phillips Family Donation and National Institutes of Health, National Center for Advancing Translational Sciences grant UL1TR000445.
Authorship
Contribution: M.R.D., M.R., and K.I.A. designed the study; S.C., D.L.G., and K.I.A. collected the data; M.R. performed the analyses; M.R.D., M.R., A.K., and K.I.A. interpreted the results; M.R.D., M.R., and K.I.A. wrote the manuscript; and S.C., D.L.G., A.K., M.R.D., M.R., P.M., and K.I.A reviewed the manuscript prior to submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Michael R. DeBaun, Division of Hematology-Oncology, Vanderbilt-Meharry Center of Excellence in Sickle Cell Disease, 2200 Children’s Way, Suite 11101, Nashville, TN 37232; e-mail: m.debaun@vumc.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal