


Abstract
The molecular causes of many inherited platelet disorders are being unraveled. Next-generation sequencing facilitates diagnosis in 30% to 50% of patients. However, interpretation of genetic variants is challenging and requires careful evaluation in the context of a patient’s phenotype. Before detailed testing is initiated, the treating physician and patient should establish an understanding of why testing is being performed and discuss potential consequences, especially before testing for variants in genes associated with an increased risk for hematologic malignancies.
Case description
A 13-year-old girl is referred to the hematologist because of severe menorrhagia and thrombocytopenia. Reduced platelet counts and easy bruising are known to affect her father (platelets, 120 × 109/L) and paternal grandmother (platelets, 90 × 109/L). No major bleeding complications have occurred in the family, other than bleeding after surgical wisdom tooth extraction in the father. Family history is otherwise uneventful. The patient has no siblings, her brother having died in a car accident 3 years ago. The girl has some hematomas 1 to 5 cm in diameter on her legs; her platelets are 115 × 109/L; mean platelet volume is 9.8 fL (normal range, 7.8-12.3 fL); hemoglobin is 95 g/L; leukocytes are 6.4 × 109/L, with normal differential. Ferritin is low at 8 ng/mL (normal range, 22-342 ng/mL). The blood film shows normal-sized well-stained platelets; light transmission aggregometry shows reduced response to low concentrations of collagen and reversible adenosine 5′-diphosphate–induced aggregation. Diagnosis of a moderate to severe platelet function defect is made, resulting in menorrhagia and subsequent iron deficiency. Her gynecologist had already prescribed hormonal contraceptives. The hematologist administers iron IV and prescribes oral iron supplementation for 6 months to treat the iron deficiency, recommends avoidance of nonsteroidal anti-inflammatory agents, and prescribes tranexamic acid for the first 3 days of menses (if not otherwise controlled by hormonal contraceptives). Six months later, at a scheduled follow-up visit, menorrhagia is well controlled by oral contraceptives (platelets, 105 × 109/L; hemoglobin, 125 g/L; ferritin, 35 ng/mL). Because of the family history, the hematologist suspects an inherited platelet defect (IPD) and recommends genetic testing to potentially identify the underlying cause. The girl’s parents give informed consent for their daughter to undergo gene panel testing. The genetic test reveals a mutation in the gene ETV6 coding for a transcription factor.1 Mutations in ETV6 are known to cause mild to moderate thrombocytopenia, a platelet function defect, and also to be associated with an increased risk for hematologic malignancies, especially during childhood, in 10% to 30% of affected families.2 Upon learning this information, the family is highly upset and wants to know what can be done to prevent leukemia. When they learn that there are no options, they make it clear that they would never have agreed to the genetic testing had they known of this potential outcome. They ask for a monthly hematologic review and blood check of her daughter. During the following weeks, the patient’s mother develops a relapse of severe depression she had developed once before after her son’s death.
Introduction
Major progress has been made in understanding IPDs.3 Here we address how IPDs can be characterized and the need to differentiate between diagnostic workup required for patient management vs a research approach to better understand the underlying causes. We also address the ethical issues of genetic testing potentially revealing a diagnosis of heightened cancer risk and thus associated with a much higher psychological disease burden than expected in the context of evaluating a mild to moderate bleeding disorder.
Platelets express >2500 different proteins.4 Therefore, IPDs can present with wide variability in severity,5 with symptoms ranging from easy bruising and menorrhagia to life-threatening bleeding during invasive procedures or injuries.6 The prevalence of individual IPDs is low, but little information exists on the overall prevalence of all IPDs combined. Recessive IPDs with a severe bleeding phenotype seem to be more frequent in societies with a high prevalence of consanguinity.7
IPDs can result in reduced platelet count (thrombocytopenia), impaired platelet function (thrombocytopathy), or both.8 Some IPDs are associated with a syndromic phenotype,9-11 because the affected proteins are also important for other cell/organ function. Another group of IPDs, caused by mutations in genes relevant for early cell differentiation,12 are associated with an increased risk for hematologic malignancies.
A recently proposed classification reflects these different scenarios6 : IPDs affecting only platelets, IPDs with a syndromic phenotype, and IPDs associated with increased risk for hematologic malignancies.
Therapeutic options for patients with IPDs
In a large majority of patients with IPDs, treatment is symptomatic. Important for diagnostic considerations, in contrast to bleeding disorders associated with plasma factor disorders, symptomatic treatment/prevention of bleeding in IPDs does not differ depending on the underlying molecular defect. Avoiding drugs inhibiting platelet function, treatment of iron deficiency, control of menorrhagia,13 tranexamic acid/desmopressin to transiently improve hemostasis,14 and platelet transfusions in cases of severe bleeding are the mainstays of treatment.15 However, in Glanzmann thrombasthenia, platelet transfusion should be avoided as long as possible to prevent induction of isoantibodies. Recombinant factor VIIa has been used successfully to control major bleeding, especially in Glanzmann thrombasthenia. Thrombopoietin receptor agonists can increase the platelet count in congenital amegakaryocytic thrombocytopenias caused by thrombopoietin mutations and transiently in others, like MYH9 disorders.16,17 Stem cell transplantation is an option in patients with amegakaryocytic thrombocytopenia, very severe platelet function defects, or syndromic disorders, in which the symptoms caused by impairment of other cells (besides platelets) are the indication for transplantation (eg, Wiskott-Aldrich syndrome).18,19
Why should IPDs be diagnosed?
IPDs need to be recognized to avoid misdiagnosis of immune thrombocytopenia. In most series of thrombocytopenic IPD patients, at least 10% of index patients are splenectomized,20 and nearly all undergo unnecessary treatment with steroids. Other patients (eg, those with ANKRD26-related thrombocytopenia) have been misdiagnosed as having myelodysplastic syndrome and received chemotherapy.21
Genetic counseling or prenatal diagnosis might be a clinical rationale for identifying the cause of an IPD. In syndromic forms, like in MYH9 disorders, which are associated with an increased risk for renal disease,22 the diagnosis can prompt regular control of renal function and treatment with renin-angiotensin system–blocking agents when proteinuria begins, which prevents progression to renal failure.23
A very different reason to identify the cause of IPDs is the scientific interest in better understanding the underlying mechanisms. With the new tools available, it is important to clearly define the reason for a more intensified diagnostic approach in patients with suspected IPDs (ie, to define clinical management vs to better understand the molecular causes).
Current diagnostic tools for IPDs
First-line diagnostic tools consist of basic techniques24 : a careful (family) history and physical examination, including a standardized bleeding score,25,26 platelet count and size (mean platelet volume),27,28 assessment of the blood film for reduced numbers of large platelets (macrothrombocytopenia), poorly stained gray platelets,29 inclusion bodies within the leukocytes, and most likely the immature platelet fraction as well.30
The second tier includes platelet function studies.31 The gold standard is light transmission aggregometry, which has several drawbacks; it requires fresh blood, at least 80 000 platelets per µL, making it problematic in thrombocytopenic patients and in patients with large platelets, which sediment with the red cell fraction during preparation of platelet-rich plasma.32 The latter can be overcome by whole-blood aggregometry.33 Lumiaggregometry is an additional tool to detect reduced-platelet dense-granule adenosine 5′-triphosphate content and/or release.34 All aggregometry tests require a relatively large blood volume, making them less suitable for pediatric patients. Functional flow cytometry and microfluidic tools require less blood but are not well standardized. Absence or reduction of surface-expressed platelet glycoproteins (eg, IIbIIIa, IbIX) are readily detected by flow cytometry.35
The third tier includes more-specialized tests like electron microscopy, western blotting, and determination of specific enzyme activities.36 Despite all these approaches and tools, unraveling the cause of an IPD remains difficult, and the methods are poorly standardized.37 Genetic testing overcomes several of these shortcomings (Figure 1).
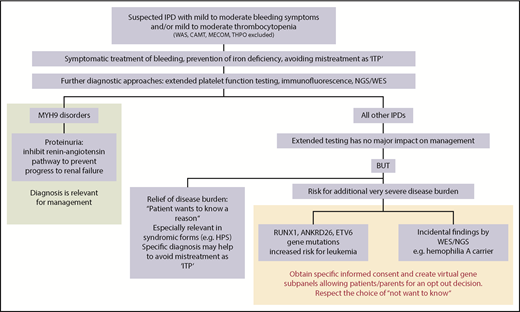
Impact of extended testing on clinical management of patients with IPDs. Once IPDs are ruled out, which may require stem cell transplantation or substitution of defect thrombopoietin, extended diagnosis of IPDs is primarily relevant for patients with MYH9 disorders. In these patients, inhibition of the renin-angiotensin pathway in case of proteinuria can prevent progression to renal failure. In all other IPDs, extended testing has no major impact on management. However, for some patients, it might be a relief of disease burden to know the reason for the bleeding tendency, but identifying the reason is often more important for concomitant disease manifestations in syndromic forms. Documentation of the specific diagnosis may also help to avoid later mistreatment as immune thrombocytopenia. It is important to be aware of the risk of causing additional severe disease burden when extended testing (either by NGS or other methods like immunofluorescence) reveals gene mutations with an increased risk for leukemia or other pathologic variants as incidental findings. At the least, specific informed consent should be obtained before testing for gene mutations with an increased risk for malignancies (RUNX1, ANKRD26, ETV6) and for reporting of incidental findings. Patients have the right to “not know.” This could be respected by creating virtual gene panels, allowing patients/parents an opt-out decision. The informed consent procedure before extended testing for IPDs, especially using NGS, should be very specific on why tests are performed, which results may be obtained, including the consequences they may have, and whether the patient wants to know results that have no direct relevance for management and/or treatment. CAMT, congenital amegakaryocytic thrombocytopenia; HPS, Hermansky-Pudlak syndrome; ITP, immune thrombocytopenia; MECOM, congenital amegakaryocytic thrombocytopenia and radioulnar synostosis; THPO, thrombopoietin mutation; WAS, Wiskott-Aldrich syndrome; WES, whole-exome sequencing.
Impact of extended testing on clinical management of patients with IPDs. Once IPDs are ruled out, which may require stem cell transplantation or substitution of defect thrombopoietin, extended diagnosis of IPDs is primarily relevant for patients with MYH9 disorders. In these patients, inhibition of the renin-angiotensin pathway in case of proteinuria can prevent progression to renal failure. In all other IPDs, extended testing has no major impact on management. However, for some patients, it might be a relief of disease burden to know the reason for the bleeding tendency, but identifying the reason is often more important for concomitant disease manifestations in syndromic forms. Documentation of the specific diagnosis may also help to avoid later mistreatment as immune thrombocytopenia. It is important to be aware of the risk of causing additional severe disease burden when extended testing (either by NGS or other methods like immunofluorescence) reveals gene mutations with an increased risk for leukemia or other pathologic variants as incidental findings. At the least, specific informed consent should be obtained before testing for gene mutations with an increased risk for malignancies (RUNX1, ANKRD26, ETV6) and for reporting of incidental findings. Patients have the right to “not know.” This could be respected by creating virtual gene panels, allowing patients/parents an opt-out decision. The informed consent procedure before extended testing for IPDs, especially using NGS, should be very specific on why tests are performed, which results may be obtained, including the consequences they may have, and whether the patient wants to know results that have no direct relevance for management and/or treatment. CAMT, congenital amegakaryocytic thrombocytopenia; HPS, Hermansky-Pudlak syndrome; ITP, immune thrombocytopenia; MECOM, congenital amegakaryocytic thrombocytopenia and radioulnar synostosis; THPO, thrombopoietin mutation; WAS, Wiskott-Aldrich syndrome; WES, whole-exome sequencing.
Genetic testing for IPDs
Next-generation sequencing (NGS) is a promising tool for diagnosis of IPDs. At least 51 genes causing IPDs have been identified, and gene panels have been established including the most common affected genes.38-45 There is enthusiasm to push for a more prominent place for NGS in the diagnostic workup for IPDs.44,46 NGS requires a small amount of blood, allowing diagnosis even in young children. DNA is stable, allowing shipment of blood samples. Costs for NGS are going down, and in comparison with the many highly specialized tests otherwise applied, NGS already seems cost effective.
However, with whole-genome sequencing, variants of unknown significance are also found. Others, like copy-number variants, are not easily detected. Taken together, in early 2019, NGS allows diagnosis in ∼50% of patients with inherited thrombocytopenia and in ∼25% of patients with inherited platelet function defects in white populations. In patients with platelet function defects, interpretation of NGS findings is particularly challenging, because defects are often caused by a composite of several variants occurring in 1 individual.47
The risk of overinterpretation of NGS findings cannot be stressed enough. Variants of unknown significance should not be implicated as a potential cause, and it is even questionable whether such variants should be shared with the patient. A variant should only be described as pathogenic if it has been described in at least 3 unrelated index patients and is reported in the Human Gene Mutation Database48 and/or the ThromboGenomics variant data set49 as a disease mutation. Variants should only be reported as likely pathogenic if they are described in 1 or 2 unrelated index patients and reported in these databases. In any case, interpretation of the clinical significance of a variant requires careful evaluation, ideally by a multidisciplinary expert team41 that interprets the genetic findings in the context of the phenotype of the patient.50,51 However, this is becoming increasingly difficult, with a disconnect between the referring physician and the specialized laboratory performing genetic testing using referred blood samples.
Phenotyping of patients and platelets
Although the phenotype is important for interpretation of genetic findings, the bleeding symptoms in IPDs are unspecific, as are most alterations observed in platelet function studies.47 In syndromic IPDs, other organ manifestations can be helpful (eg, deafness/cataracts in MYH9 disorders52 ). In a collaborative approach, several groups have developed a method based on immunofluorescence staining of platelet proteins on a blood smear, which allows detailed phenotyping of platelets with a small amount of blood, applicable in young children and thrombocytopenic patients.53 It allows diagnosis of several IPDs by staining for direct or indirect markers (Glanzmann thrombasthenia, biallelic Bernard-Soulier syndrome, MYH9 disorders, gray platelet syndrome, GFI-1b disorders, β1-tubulin and α-tubulin defects, dense-granule and α-granule deficiencies, and ETV6 mutation–related IPDs).
Challenges: IPDs associated with an increased risk for malignancies
A difficult and even ethical issue is testing for IPDs associated with an increased risk for hematologic malignancies (eg, mutations in RUNX1,54 ETV6,1 and ANKRD2655,56 ). Patients with these IPDs generally present with mild to moderate bleeding symptoms and moderately decreased platelet counts, rarely causing distress for the patients or families. In contrast, the information that a mutation has been identified that is potentially associated with an increased risk for leukemia may cause major disease burden. In addition, only a subgroup of individuals with these mutations (10%-30%) will develop leukemia, and the individual risk of breakthrough is largely unknown. The situation might be somewhat different in families with inherited platelet disorders, where ≥1 members have already developed hematologic malignancies. However, for a patient with 1 of those IPDs who has developed a hematologic malignancy, genetic testing of potential related stem cell donors is important. Relatives with the same mutation should not be used as stem cell donors.57
Although ethical guidelines58 recommend genetic testing only in the context of genetic counseling, in practice most physicians referring patients’ DNA or blood smears for more specialized testing are unaware that some IPDs are associated with an increased risk of leukemia. NGS can provide incidental findings (eg, identifying a girl as a carrier of hemophilia A). Standard informed consent forms for NGS give the patient the option to decide on whether he or she wants to know additional pathologic variants.59 However, this is impossible when the same genetic variant causes the IPD and an increased risk for malignancies. For other methods that could identify these IPDs (eg, immunofluorescence), usually no specific informed consent is obtained. In addition, guidelines from several genetic societies recommend deferring genetic testing until a child is mature enough to participate in decision making,58 and physicians should respect the right of the patient to “not know.” These aspects are especially relevant in the testing of children. Responsible handling of legal regulations may become complex when patient samples are sent for genetic testing to a reference laboratory in another country or jurisdiction. Depending on the society, the health care system, or the individual family, genetic findings may create insurance problems for the patient or even discrimination. In this context, data security and confidentiality are additional highly relevant issues. Detailed discussion of all these aspects is beyond the scope of this article, but additional information can be found elsewhere.60-64
Conclusion
The molecular causes of many IPDs have been unraveled. NGS facilitates the correct diagnosis in a substantial proportion of patients. However, in IPDs, the specific diagnosis rarely affects management. The new tools do not make it any easier to comply with the responsibility toward patients in the highly sensitive field of inherited disease, neither for the clinical hematologist nor for the laboratory expert. The disconnect between patient/referring physician and testing laboratory, often hundreds of miles apart and possibly even in different jurisdictions, generates new issues related to informed consent. Interpretation of genetic variants is still challenging and requires careful evaluation by a multidisciplinary expert team in the context of a patient’s phenotype. Immunofluorescence analysis of blood smears facilitates platelet phenotype characterization in IPDs (Figure 1). Before detailed testing for identification of the underlying cause of an IPD is initiated, the treating physician and the patient should establish an understanding of why testing is performed and discuss potential consequences. This is especially important for testing of minors and before patients are tested for genetic variants in genes, potentially associated with other organ manifestations or with an increased risk for hematologic malignancies. Organizational structures allowing (anonymized) research on patients with IPDs, independent of clinical management, are urgently needed.
Acknowledgments
The authors thank the colleagues referring patients and other researchers who perform genetic testing for IPDs (especially Kathleen Freson, University Leuven, Leuven, Belgium) for valuable discussions on the ethical challenges of patient testing in IPDs. The work of Marcel Baschin and Carmen Blumentritt, who perform the immunofluorescence analysis of platelets in the Greifswald laboratory, is highly appreciated.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB TR240).
Authorship
Contribution: A.G. and J.J.M.E. drafted the concept and wrote the manuscript.
Conflict-of-interest disclosure: The authors perform platelet function testing and immunofluorescence testing for diagnosing IPDs. The authors declare no other competing financial interests.
Correspondence: Andreas Greinacher, Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, Sauerbruchstraße, 17475 Greifswald, Germany; e-mail: greinach@uni-greifswald.de.
