

Key Points
Headaches, lethargy, and abdominal pain are recurring features of nonovert cTTP disease and are highly responsive to prophylactic therapy.
The incidence of cerebrovascular events is significantly lower in cTTP patients on regular prophylactic therapy.

Abstract
Congenital thrombotic thrombocytopenic purpura (cTTP) is an ultra-rare thrombomicroangiopathy caused by an inherited deficiency of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13). There are limited data on genotype-phenotype correlation; there is no consensus on treatment. We reviewed the largest cohort of cTTP cases, diagnosed in the United Kingdom, over the past 15 years. Seventy-three cases of cTTP were diagnosed, confirmed by genetic analysis. Ninety-three percent were alive at the time of review. Thirty-six percent had homozygous mutations; 64% had compound heterozygous mutations. Two presentation peaks were seen: childhood (median diagnosis age, 3.5 years) and adulthood, typically related to pregnancy (median diagnosis age, 31 years). Genetic mutations differed by age of onset with prespacer mutations more likely to be associated with childhood onset (P = .0011). Sixty-nine percent of adult presentations were associated with pregnancy. Fresh-frozen plasma (FFP) and intermediate purity factor VIII concentrate were used as treatment. Eighty-eight percent of patients with normal blood counts, but with headaches, lethargy, or abdominal pain, reported symptom resolution with prophylactic therapy. The most common currently used regimen of 3-weekly FFP proved insufficient for 70% of patients and weekly or fortnightly infusions were required. Stroke incidence was significantly reduced in patients receiving prophylactic therapy (2% vs 17%; P = .04). Long-term, there is a risk of end-organ damage, seen in 75% of patients with late diagnosis of cTTP. In conclusion, prespacer mutations are associated with earlier development of cTTP symptoms. Prophylactic ADAMTS13 replacement decreases the risk of end-organ damage such as ischemic stroke and resolved previously unrecognized symptoms in patients with nonovert disease.
Introduction
Congenital thrombotic thrombocytopenic purpura (cTTP), also known as Upshaw-Schulman syndrome, is an ultra-rare thrombomicroangiopathy due to an inherited deficiency of the von Willebrand factor (VWF)-cleaving metalloprotease, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13), resulting in the abnormal presence of ultra-large VWF multimers and the presumed subsequent formation of circulating platelet-rich microthrombi.1 The incidence of immune thrombotic thrombocytopenic purpura (TTP) and cTTP combined has been estimated at 2 to 6 people per million per year; cTTP accounts for 2% to 10% of all TTP cases in international registries.2-4 Although first symptom occurrence has classically been described in childhood,5,6 presentation can occur at any age, with a notable peak in women during pregnancy.7,8
Disease-causing mutations in cTTP occur throughout the ADAMTS13 gene, located on chromosome 9q34.9 The ADAMTS13 gene (Figure 1) consists of 29 exons10 encompassing 1427 amino acids, and 3 main categories of disease-causing mutations are seen: frameshift, missense, and nonsense mutations. To date, over 150 mutations have been described,11 spread across all exons. Two mutations are particularly significant for their characteristic presentation: the exon 24 mutation p.R1060W (rs142572218), a single-nucleotide substitution associated with first presentation in pregnancy,8,12,13 and the exon 29 mutation c.4143dupA (rs387906343), an aberrant nucleotide insertion mutation with resulting frameshift, distinguished by its frequency in individuals of central European, Polish, and Scandinavian ancestry.14,15 However, these are notable exceptions and further understanding is required on the correlation between cTTP genotypes and the clinical phenotype.
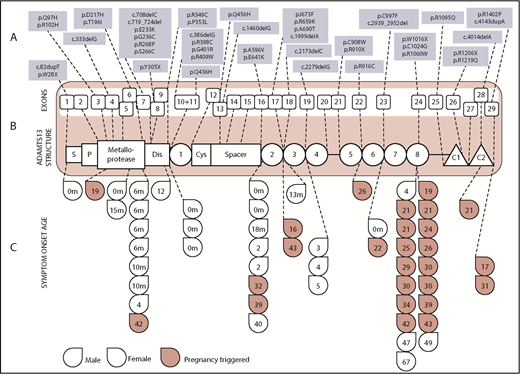
Disease-causing mutations were spread along the length of the ADAMTS13 gene. (B) The structure of the ADAMTS13 protein is shown in the red box in the middle of the diagram along with the 29 exons that make up the ADAMTS13 gene encoding for it. (A) The top third of the diagram shows all of the ADAMTS13 variations seen by exon, with novel mutations denoted in bold text. (C) The bottom third shows the age of symptom onset according to mutation location. Red drops denote those presenting in pregnancy. Only patients with mutations confined to a single exon are included. C1 and C2, type 1 and 2 CUB domains; Cys, cysteine rich; Dis, disintegrin; P, propeptide; S, signal peptide. Adapted from Zheng et al.10
Disease-causing mutations were spread along the length of the ADAMTS13 gene. (B) The structure of the ADAMTS13 protein is shown in the red box in the middle of the diagram along with the 29 exons that make up the ADAMTS13 gene encoding for it. (A) The top third of the diagram shows all of the ADAMTS13 variations seen by exon, with novel mutations denoted in bold text. (C) The bottom third shows the age of symptom onset according to mutation location. Red drops denote those presenting in pregnancy. Only patients with mutations confined to a single exon are included. C1 and C2, type 1 and 2 CUB domains; Cys, cysteine rich; Dis, disintegrin; P, propeptide; S, signal peptide. Adapted from Zheng et al.10
Treatment in cTTP is focused on replenishing ADAMTS13 levels. This can be achieved using therapeutic plasma exchange but this is rarely required as an ongoing treatment approach. Instead, infusion of ADAMTS13-rich blood products is more frequently used and, although a number of products including cryosupernatant,16 whole blood, and cryoprecipitate have historically been used,17 the most common treatment is fresh-frozen plasma (FFP) infusion.18 In the United Kingdom, intermediate purity factor VIII concentrate (BPL 8Y) is also used,19,20 a virally inactivated plasma-derived factor VIII product containing ADAMTS1321 with lower risks of fluid overload and allergic reactions, and the potential for patients to self-administer. Furthermore, FFP used in the United Kingdom is non–United Kingdom sourced, having undergone double viral inactivation and prion reduction steps.22-24 Although there is limited evidence on the risk of end-organ damage in cTTP,25 the incidence of this may be underestimated due to patients dying without the diagnosis being made.26 Additionally, little has been published on the significance of ongoing symptoms experienced by patients with this condition. As a result, consensus on treatment frequency, as well as on whether these chronically ADAMTS13-deficient individuals require regular maintenance therapy or only ad hoc treatment at high-risk periods such as pregnancy and surgery, is lacking.
We undertook a review of all of our cases of cTTP to evaluate the potential impact of the genotype and differing treatment regimens on short-term and longer-term disease outcomes.
Method
Since January 2009, the UK TTP registry has been collecting information on all acute presentations of TTP across the country. Although patients were primarily taken from the registry, additional cases from before its formation were also identified from its precursor, the South East England Registry for TTP.2
All cases of confirmed TTP are consented to the UK TTP Registry (Multicentre Research Ethics Committee [MREC]: 08/H0810/54). Data collected by the registry include demographic information, ADAMTS13 activity, and laboratory and organ damage markers at acute presentation and subsequent follow-up. cTTP was considered in patients who presented with thrombocytopenia and microangiopathic hemolytic anemia (MAHA) with ADAMTS13 protease activity below 10 IU/dL (fluorescence resonance energy transfer substrate VWF73 [FRETS VWF73] assay; normal range, 60% to 146%) and no evidence of anti-ADAMTS13 immunoglobulin G (IgG) antibodies (normal range, <6%). A diagnosis of cTTP was defined as fulfillment of these criteria in addition to confirmation of the presence of a cTTP-linked genetic mutation (either a homozygous mutation or 2 heterozygous mutations)27 (compound heterozygous). In this observational study, once a diagnosis of cTTP was confirmed, treatment decisions were at the discretion of the physician caring for the patient. A follow-up questionnaire was distributed to retrospectively evaluate treatment course and clinical outcomes.
The genomic database Ensembl28 was used alongside a literature search of PubMed and Google Scholar to identify known cTTP mutations. For novel or previously unreported mutations, PolyPhen-229 was used to assess the likelihood of a mutation being disease causing. Additionally, the diagnosis was also considered in individuals with first-degree relatives known to have a diagnosis of cTTP. Such individuals satisfied the same diagnostic criteria but were often preemptively tested on the basis of family history and so did not necessarily present with symptoms of TTP, thrombocytopenia, and/or MAHA.
Citrated blood samples for ADAMTS13 activity, anti-ADAMTS13 IgG antibodies, and ADAMTS13 antigen were taken at first presentation. For patients who presented with an acute TTP event and required therapeutic plasma exchange, blood samples were taken prior to the first exchange and repeated in remission and at follow-up to confirm persisting ADAMTS13 deficiency and the absence of anti-ADAMTS13 antibodies. EDTA whole blood was taken for genetic testing once a diagnosis of cTTP was suspected. ADAMTS13 activity was measured using the FRETS VWF73 method30 and previously published enzyme-linked immunosorbent assay techniques were used for quantification of the anti-ADAMTS13 IgG antibodies31,32 and ADAMTS13 antigen.33 Genetic mutations were identified using Sanger sequencing, prior to which the 29 ADAMTS13 exons were amplified using a polymerase chain reaction method and custom oligonucleotide primers. National Center for Biotechnology Information (NCBI) NG_011934.234 was used as the reference sequence. DNA-sequencing analysis was undertaken both manually and using Sequencher 5.4.6 (Gene Codes Corporation, Ann Arbor, MI).
All patients with a cTTP diagnosis confirmed by genetic analysis were included in the statistical analysis with the χ2 test, Student t test, Mann-Whitney U test, or Kruskal-Wallis test used as appropriate. Graphpad Prism 6 (GraphPad Software Inc, La Jolla, CA) was used for all statistical analyses.
Results
In addition to 819 TTP patients from the UK TTP Registry, 222 patients from its predecessor, the South East England registry, were included. Therefore, a total of 1041 patients presenting from 2003 to 2018 were included in the study. Of these 1041 patients, 100 were suspected to have cTTP and underwent genetic testing. Seventy-three patients from 66 families were found to have a cTTP-causing mutation. Seventy percent (n = 51) of cTTP patients were female. Ninety-three percent of patients (n = 68) were alive at the time of review with 4 of the 5 patients dying as a result of a stroke and 1, unrelated to TTP, from cancer. The median length of follow-up was 100 months (range, 11-482 months).
Two peaks in presentation were seen: early childhood and during pregnancy. Eighty-six percent of males presented in the neonatal period or childhood (childhood-onset cTTP) and 61% of females presented in pregnancy. Childhood-onset cTTP accounted for 38% of all cases (n = 28) with a median age of diagnosis of 3 years (range, <1 month to 12 years). The most common diagnostic prompt was thrombocytopenia, seen in 100% of patients, with MAHA present in 29% of cases (n = 9). The most common precipitant of childhood-onset cTTP was infection, present in 40% of cases (n = 11). Within the cohort of childhood-onset cases were 4 patients who presented in the neonatal period with hyperbilirubinemia and thrombocytopenia. The median age of diagnosis when considering all adult-onset disease was 31 years (range, 16-71 years). Pregnancy was the most common trigger of adult-onset cTTP, accounting for 69% of adult presentations (n = 31). MAHA was the diagnostic presentation in 90% of patients (n = 23) with the remaining patients being diagnosed following investigation for thrombocytopenia. Forty-three percent of patients presenting in childhood (n = 12) were born by consanguineous parents compared with 6.7% of patients presenting in adulthood (n = 3). Three West African–origin patients presented in childhood; this is an ethnic group in whom cTTP has not previously been reported. Demographic variations are listed in Table 1.
Demographics according to period of presentation
| Onset period | No. (% total) | Female, % | Median symptom onset, age (range) | Median diagnosis, age (range) | Consang, % | Ethnicity, % |
|---|---|---|---|---|---|---|
| Neonatal/childhood | 28 (38) | 32 | 10 mo (<1 mo to 12 y) | 3.5 y (<1 mo to 12 y) | 43 | White, 39; S Asian, 36; Arab, 14; African, 11 |
| Pregnancy | 31 (42) | 100 | 26.5 y (16 y to 42 y) | 29 y (16 y to 43 y) | 7 | White, 87; S Asian, 10; SE Asian, 3 |
| Late onset | 12 (17) | 75 | 36 y (16 y to 67 y) | 56 y (27 y to 71 y) | 8 | White, 92; S Asian, 8 |
| Family history | 2 (3) | 100 | 3.5 y (0 mo to 7 y) | 26.5 y (1 y to 50 y) | 0 | White, 100 |
| Onset period | No. (% total) | Female, % | Median symptom onset, age (range) | Median diagnosis, age (range) | Consang, % | Ethnicity, % |
|---|---|---|---|---|---|---|
| Neonatal/childhood | 28 (38) | 32 | 10 mo (<1 mo to 12 y) | 3.5 y (<1 mo to 12 y) | 43 | White, 39; S Asian, 36; Arab, 14; African, 11 |
| Pregnancy | 31 (42) | 100 | 26.5 y (16 y to 42 y) | 29 y (16 y to 43 y) | 7 | White, 87; S Asian, 10; SE Asian, 3 |
| Late onset | 12 (17) | 75 | 36 y (16 y to 67 y) | 56 y (27 y to 71 y) | 8 | White, 92; S Asian, 8 |
| Family history | 2 (3) | 100 | 3.5 y (0 mo to 7 y) | 26.5 y (1 y to 50 y) | 0 | White, 100 |
Neonatal and childhood presentations were more likely to be associated with consanguinity and accounted for all cases of cTTP seen in patients of African or Middle Eastern ethnic origin. Pregnancy-associated presentations were most commonly associated with the exon 24 rs142572218 mutation.
Consang, consanguineous; S Asian, south Asian; SE Asian, southeast Asian.
Genetic analysis
Thirty-six percent of cTTP patients (n = 26) had homozygous mutations; 64% (n = 47) had compound heterozygous mutations. The most common mutation seen was the exon 24 R1060W missense mutation (rs142572218; n = 11 homozygous, 21 heterozygous), associated with later-onset disease presentation.
A clear correlation was seen between the mutation location and the age at presentation. When considering the 26 patients with homozygous mutations, the median age of diagnosis with prespacer mutations was 24 months, compared with 294 months for postspacer mutations (P < .0001). Ten percent of adult presentations were associated with prespacer mutations compared with 52% of childhood presentations (P = .0011). Of the remaining 17 compound heterozygotes patients with both pre- and postspacer mutations, 14 presented in adulthood and 3 in childhood. Twelve of the adult presentations were in pregnancy, 9 of which were found to be heterozygous for the exon 24 R1060W missense mutation (rs142572218).
Symptoms and treatment
Regular prophylactic treatment was commenced in 67% of patients (n = 49), 12% (n = 9) required on-demand treatment of cTTP-precipitating events such as infection or pregnancy, and 21% of patients (n = 15) received no prophylactic treatment excluding their initial acute presentation episode (Table 2). In initially untreated patients, the symptoms most frequently reported were recurrent headaches and nonhemiplegic migraines (experienced by 44% of patients; n = 32), lethargy (18%; n = 13), and abdominal pain (14%; n = 10). Eight percent of cTTP had experienced transient ischemic attack (TIAs; n = 6) and 19% experienced strokes (n = 14), either at presentation or following diagnosis. Urine protein-to-creatinine ratio (PCR; normal range, 0-13 mg/mmol) was checked in 50 of the 73 patients and was raised in 20% (n = 10) with a median age of 24 years (range, 13-40 years) at a time of the raised PCR being measured. Eight of these 10 patients commenced regular prophylactic replacement therapy with resolution of the proteinuria. Within the whole cohort, 3 patients had a permanent reduction in renal function: 1 with a childhood diagnosis of cTTP who did not commence prophylactic therapy until end-stage renal failure requiring renal transplant had occurred; 1 who was diagnosed with cTTP in her seventh decade; and 1 in whom proteinuria was noted during her fourth pregnancy, 6 years after the initial cTTP diagnosis. Regular FFP was commenced during this pregnancy and ongoing thereafter but proteinuria persisted postpartum.
Treatment type and frequency by age
| Age, y | Prophylactic FFP | Prophylactic BPL 8Y | On demand | No treatment |
|---|---|---|---|---|
| <16 (n = 17) | n = 0 | n = 16 | n = 1 | n = 0 |
| Once/wk, n = 5 | ||||
| Every 10 d, n = 1 | ||||
| Every 2 wk, n = 6 | ||||
| Every 3 wk, n = 2 | ||||
| Every 4 wk, n = 2 | ||||
| ≥16 (n = 56) | n = 23 | n = 10 | n = 8 | n = 15 |
| Three/wk, n = 1 | Twice/wk, n = 6 | |||
| Once/wk, n = 5 | Once/wk, n = 4 | |||
| Every 2 wk, n = 11 | ||||
| Every 3 wk, n = 6 | ||||
| Every month, n = 1 |
| Age, y | Prophylactic FFP | Prophylactic BPL 8Y | On demand | No treatment |
|---|---|---|---|---|
| <16 (n = 17) | n = 0 | n = 16 | n = 1 | n = 0 |
| Once/wk, n = 5 | ||||
| Every 10 d, n = 1 | ||||
| Every 2 wk, n = 6 | ||||
| Every 3 wk, n = 2 | ||||
| Every 4 wk, n = 2 | ||||
| ≥16 (n = 56) | n = 23 | n = 10 | n = 8 | n = 15 |
| Three/wk, n = 1 | Twice/wk, n = 6 | |||
| Once/wk, n = 5 | Once/wk, n = 4 | |||
| Every 2 wk, n = 11 | ||||
| Every 3 wk, n = 6 | ||||
| Every month, n = 1 |
Patients either received regular prophylactic FFP or BPL 8Y or no regular prophylactic treatment. For patients not on prophylaxis, on-demand treatment was given during events precipitating a cTTP exacerbation such as pregnancy or infection. The on-demand column signifies patients who required on-demand treatment and the no-treatment column patients who had never needed or received treatment since their initial presenting event. The child who needed on-demand therapy received BPL 8Y whereas all 8 adults who needed on-demand treatment were given FFP.
Treatment was instigated at the discretion of the treating physician. FFP (Octaplas; Octapharma, Vienna, Austria) was given at a dose of 10 to 15 mL/kg 3 weekly initially and BPL 8Y 10 to 20 IU/kg with a starting regimen of 1 treatment every fortnight. Treatment frequency was then increased or decreased according to patient symptoms and, where applicable, platelet counts. For children and adolescents, the most common reason for increasing treatment frequency was thrombocytopenia. For adults, the most common reason was nonovert symptoms such as persisting headaches or lethargy. BPL 8Y doses were increased until symptoms had resolved or a maximum dose of 20 IU/kg was reached. For FFP, treatment was increased in frequency in a stepwise manner up to once weekly with the exception of 1 patient who was increased to infusions 3 times a week due to persisting symptoms. Factor VIII:C levels were not measured routinely in patients receiving BPL 8Y but for, in light of previous studies showing increased levels in the first 24 hours postadministration,19 a single dose of prophylactic low-molecular-weight heparin thromboprophylaxis sometimes given on the day of BPL 8Y administration depending on the treating center. Sixty-seven percent of patients received regular prophylactic replacement therapy, 12% received on-demand therapy, and 21% had never received therapy since the initial diagnosis of cTTP. Regular prophylactic treatment was more commonly used in children with 94% of patients under 18 years of age on regular therapy in comparison with 61% of those over the age of 18 years. BPL 8Y was used as a source of ADAMTS13 in 100% of children treated prophylactically. Sixty-four percent of adults treated prophylactically received FFP infusions and 36% received BPL 8Y. Of the 23 adults who received prophylactic FFP treatment, 70% (n = 16) required an increase in treatment frequency due to insufficient symptom control with 3 weekly infusions. Treatment frequencies are detailed in Table 2. Choice of prophylactic replacement therapy to commence was at the discretion of the treating physician. At the time of review, 14% of patients (n = 10) had previously switched between BPL 8Y and FFP. Three adults switched from BPL 8Y to FFP as a result of inadequate control of their symptoms despite a normal platelet count and with subsequent complete symptom resolution upon switching. Seven patients switched from FFP to BPL 8Y: 1 patient due to severe reactions to plasma infusion, 1 due to patient preference, and 5 children (in line with national experience of BPL 8Y use in children). No patients developed inhibitors with either FFP or BPL 8Y treatment and no new ADAMTS13 antibodies were detected.
Of the patients receiving regular prophylactic treatment, 23% were commenced due to thrombocytopenia; 66% due to symptoms such as lethargy, headaches, and abdominal pain, despite a normal platelet count; and 11% as primary prophylaxis, predominately siblings of previously known cTTP patients. Patients experienced significant resolution of symptoms with commencement of regular prophylactic therapy as summarized in Table 3. All patients treated for abdominal pain, lethargy, or headaches (n = 24) had a normal platelet count at commencement of prophylaxis. Thirty-three percent of these patients (n = 8) were followed up with weekly blood counts and the median platelet count in the 2- to 3-month period posttreatment commencement had increased (median preprophylaxis, 261 × 109/L; median postprophylaxis, 325 × 109/L; P = .23). The use of prophylactic replacement therapy appeared to reduce the incidence of stroke. Two percent of the cohort (n = 1) had a stroke after starting regular prophylactic treatment in comparison with 17% (n = 4) of patients not on regular therapy (P = .04) during the follow-up period. Of the 4 adults who died due to complications of cTTP, 1 was receiving 3-weekly prophylactic FFP infusions and 3 were on no replacement treatment.
Signs and symptoms leading to the commencement of regular prophylactic therapy
| Symptom | Patients affected, % | Patients whose symptoms resolved with therapy, % |
|---|---|---|
| Abdominal pain | 9 (n = 4) | 100 |
| Headaches and migraines | 23 (n = 11) | 82 (n = 9) |
| Lethargy | 19 (n = 9) | 89 (n = 8) |
| Persistent thrombocytopenia | 23 (n = 11) | 91 (n = 10) |
| Recurrent MAHA episodes | 4 (n = 2) | 100 |
| Stroke and TIAs | 11 (n = 5) | 80 (n = 4) |
| Symptom | Patients affected, % | Patients whose symptoms resolved with therapy, % |
|---|---|---|
| Abdominal pain | 9 (n = 4) | 100 |
| Headaches and migraines | 23 (n = 11) | 82 (n = 9) |
| Lethargy | 19 (n = 9) | 89 (n = 8) |
| Persistent thrombocytopenia | 23 (n = 11) | 91 (n = 10) |
| Recurrent MAHA episodes | 4 (n = 2) | 100 |
| Stroke and TIAs | 11 (n = 5) | 80 (n = 4) |
Late recognition of cTTP
Patient histories were retrospectively reviewed for evidence of prior clear TTP symptoms including unexplained MAHA, thrombocytopenia, or anemia during illness or pregnancy; early TIAs or stroke; and otherwise unexplainable obstetric complications. Fifty-one percent of patients (n = 37) were diagnosed with cTTP at the time of first symptom onset. Of the patients who were not diagnosed at first presentation, the median period between first symptoms and correct diagnosis was 47 months (range, 2-516 months.). Patients with a delayed diagnosis were erroneously labeled as having chronic immune thrombocytopenic purpura, immune-mediated TTP, and Evans syndrome prior to the correct diagnosis of cTTP being made. Medically significant complications encountered prior to diagnosis are listed in Table 4.
Complications seen prior to cTTP diagnosis in adult patients for which no other clear causative factor was found
| Complication | No. (% of total) |
|---|---|
| Facial palsy | 2 (2.8) |
| Miscarriage/stillbirth* | 8 (11) |
| Pulmonary hemorrhage | 1 (1.4) |
| Retinal vein thrombosis | 1 (1.4) |
| Seizures | 1 (1.4) |
| Stroke/TIA* | 18 (25) |
| Third-trimester pregnancy complications excluding miscarriage* | 4 (5.6) |
| Visual defects | 3 (4.2) |
| Complication | No. (% of total) |
|---|---|
| Facial palsy | 2 (2.8) |
| Miscarriage/stillbirth* | 8 (11) |
| Pulmonary hemorrhage | 1 (1.4) |
| Retinal vein thrombosis | 1 (1.4) |
| Seizures | 1 (1.4) |
| Stroke/TIA* | 18 (25) |
| Third-trimester pregnancy complications excluding miscarriage* | 4 (5.6) |
| Visual defects | 3 (4.2) |
The number of patients who had miscarriages or stroke/TIAs rather than the total number of events; third-trimester complications seen were antepartum hemorrhage, fetal distress, placental abruption, and preeclampsia.
Late recognition of cTTP and treatment commencement was associated with adverse outcomes. Eight patients (11% total) were diagnosed with cTTP at a median age of 60 years (range, 43-71 years). Two of these patients did not experience major cTTP sequelae but of the remaining 6, 1 was found to have marked renal and cardiac dysfunction and 5 had experienced at least 1 stroke by 50 years of age. In 2 of these patients, stroke was the cause of death. Four of the 6 female patients gave clear histories of prior strokes or miscarriages during pregnancy with associated thrombocytopenia.
Discussion
As is often the case in ultra-rare disorders, there is limited evidence on the outcomes or most appropriate management for patients with cTTP. Increased awareness of this disorder and availability of ADAMTS13 assays is likely to result in an increase in the diagnosis of cTTP, and, when coupled with recombinant ADAMTS13 therapy (which is on the horizon),35 this mandates greater understanding of and clarity into the scientific basis and the clinical outcomes in this disease. This is the largest study correlating both the genotypic and phenotypic features and outcomes of replacement therapy in patients with cTTP, utilizing 1 of the largest TTP registries internationally. We report 7 new mutations and include 3 patients of West African origin who, to our knowledge, are the first non-Arabic African36 ethnicity patients reported to have cTTP. We also report similar findings to previous studies in cTTP that showed childhood and pregnancy peaks of presentation37,38 and found that mutations earlier in the ADAMTS13 sequence were potentially associated with earlier age of onset than later mutations.39,40 The reiteration of this correlation between prespacer gene mutations and presenting age is a finding with potential clinical relevance as it suggests that patients with prespacer mutations potentially have more severe disease and so should be considered for earlier prophylactic therapy. A further meaningful finding is that over 10% (n = 8) of the patients in this study were first diagnosed with cTTP between the ages of 43 and 71 years of age. Although conventional wisdom is that it is extremely unlikely for a patient with cTTP to first present beyond early adulthood, this is not dissimilar from the next largest cTTP study,41 which found 9% of Japanese patients being diagnosed between 45 and 63 years of age. These are not insignificant proportions and suggest that in adult patients of all ages, cTTP should be considered a diagnosis if there is a thrombocytopenia and unexplained neurological or thrombotic events.
The most important finding of this study is the identification of a cohort of patients with nonovert symptoms responding to ADAMTS13 replacement therapy. Of the 24 patients commenced on regular prophylactic therapy for headaches, lethargy, and abdominal pain, 88% reported symptom resolution with therapy. All of these patients had normal blood counts with no thrombocytopenia, normal lactate dehydrogenase levels, and no evidence of end-organ damage. Interestingly, although statistical significance was not conclusively proved, there is also a suggestion that the median platelet count for these patients increased with prophylaxis. These findings likely suggest that ongoing nonovert disease is present in these patients. The relevance of this may be linked to our discovery that late recognition of cTTP was found to lead to poor outcomes, associated with significant end-organ damage, although the overall patient numbers are small in this group. Although there are no published long-term data on the effects of cTTP, these findings suggest that patients with cTTP can suffer from overt episodes of disease that are associated not only with classical features including MAHA and thrombocytopenia but also with symptoms when the condition is not overt. It is likely that small-platelet VWF aggregates can occlude the microcirculation, causing symptoms, without producing sufficient platelet consumption to result in the development of thrombocytopenia. The end-organ damage seen in older patients may be partially explained by the increase in VWF with age,42 and this increase in VWF may similarly explain the excess of presentations of women with cTTP in pregnancy. The finding of raised urinary PCR further suggests that small-vessel or endothelial damage mediated by small-platelet VWF aggregates may occur in the absence of overt features of disease. Given these findings, we would suggest that at the very least, patients with cTTP who are symptomatic, if not all patients, should be considered for regular prophylactic therapy to prevent early morbidity regardless of a normal platelet count or overt evidence of end-organ damage.
We would currently suggest FFP as the preferred prophylactic option as studies43 suggest that it achieves higher posttreatment ADAMTS13 activity levels than BPL 8Y19,20 and, internationally, it is more widely available than BPL 8Y or other intermediate purity factor VIII concentrates such as Koate-DVI44 ; however, it should be noted that no patients on BPL 8Y reported adverse effects. What has not been investigated in detail in this study or, subject to consensus, is the dose, frequency, or indeed the target peak and trough level of ADAMTS13 required for adequate prophylactic therapy. However, this study has provided important not previously documented observations. Plasma was typically given at 10 to 15 mL/kg initially at 3-weekly intervals based on current consensus,37,45-48 increasing up to weekly dependent on the response of the platelet count or symptom improvement. Despite this being the most common treatment regimen used internationally, it should be noted that this was found insufficient to fully control symptoms in 70% of patients. It is likely that this is a result of current protocols underestimating the significance of the aforementioned nonovert symptoms and focusing on normalization of platelet counts as a marker of treatment response. Although 87% of patients (n = 21) had normal platelet counts with 3 weekly plasma infusions, only 1 patient being treated for nonovert symptoms found this schedule sufficient to alleviate symptoms. Our suggestion would be that patients commence on 10 to 15 mg/kg FFP at fortnightly intervals with a low threshold to increase to weekly infusions should this fail to alleviate any symptoms. Although thrombocytopenia should be considered as a trigger to increase treatment frequency, a normal platelet count should not be taken to mean treatment is sufficient; instead, clinical symptoms and markers of organ damage such as a rising urine PCR should be considered.
It should be noted, however, that the rates of FFP reactions seen in this study are likely lower than the international average given the widespread use of pooled, solvent-detergent FFP and its suggested lower risk of adverse events when compared with standard FFP.49 In some patients, frequent reactions to FFP or the inability to tolerate large volumes of plasma will limit the ability to treat satisfactorily, reinforcing the need for a recombinant ADAMTS13 replacement product for such individuals. In the interim, we suggest patients intolerant of FFP be treated with an intermediate purity factor VIII concentrate.
There are limitations to this study: although the number of patients included surpasses any previous work on cTTP, the numbers are still small and treatment pathways have not been part of a formal study. This is particularly relevant for factors such as assessing whether treatment reduces the risk of stroke, which should ideally be undertaken in a prospective study. However, these numbers have been gathered as part of a registry, which provides important conclusions in ultra-rare diseases. Only collaborative international projects will be able to address this more definitively. Additionally, data have been collected both prospectively and retrospectively with the latter introducing the risk of recall bias. It should also be noted that the rarity of the condition and the lack of treatment consensus means that there is a wide range of treatment given, which compromises the ability to easily compare groups. This is something that should improve with great evidence of successful treatment approaches. Finally, this study does not include patients who may have presented with and died of cTTP before the diagnosis was made or the patient was consented for the UK TTP Registry.
In conclusion, we report that prespacer mutations are associated with earlier development of cTTP symptoms. A nonovert clinical picture exists in patients with cTTP with symptoms including lethargy, headaches, and abdominal pain occurring in the absence of typical clinical and laboratory features of active TTP. Prophylactic treatment is associated with significant improvement in these symptoms and long-term outcomes relating to organ failure. Although identification of nonovert symptoms has been commented on in previous published cases, this study confirms their frequency and the benefit of treatment. Routine laboratory parameters are not sensitive in identifying patients at risk of complications of cTTP and, as such, therapy should be considered for symptomatic patients to prevent early morbidity and mortality.
Please contact ferras.alwan@nhs.net for original data requests.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
F.A. was supported by unrestricted educational funding from Shire.
Authorship
Contribution: F.A. wrote the paper, collected data, and undertook laboratory testing; C.V., R.L., A.C., W.L., T.D., W.T., R.G., T.B., H.G.W., N.C., R.R., T.C., J.J.v.V., Q.A.H., S.D., J.M., N.B., N.P., M.D., J.A., M.P.C., H.L., J.-P.W., and M.T. collected data and reviewed the manuscript; and M.S. was senior author, collected data, and reviewed the manuscript.
Conflict-of-interest disclosure: A.C., J.J.v.V., and R.G. received honoraria from Ablynx for attendance at advisory boards and speaker fees from Alexion. M.S. received honoraria from Ablynx, Alexion, Novartis, and Shire for attendance at advisory boards and speaker fees from Octapharma. M.T. received honoraria from Ablynx for attendance at advisory boards. Q.A.H. received speaker fees from Shire. R.L. received honoraria from Bayer, Roche, Octapharma, Shire, and Grifols for attendance at advisory boards and speaker fees from Octapharma, SOBI, Novo Nordisk, Bayer, and Bio Products Laboratory. T.D. received honoraria from Ablynx and Alexion for attendance at advisory boards and speaker fees from Alexion, Roche and Shire. W.L. received honoraria from Ablynx and Alexion for attendance at advisory boards. W.T. received honoraria from Ablynx for attendance at advisory boards and funding from Octapharma to attend an educational meeting. The remaining authors declare no competing financial interests.
Correspondence: Ferras Alwan, Department of Haematology, University College London Hospital, 250 Euston Rd, London NW1 2BU, United Kingdom; e-mail: ferras.alwan@nhs.net.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal