

Abstract
Allogeneic Hematopoietic Stem Cell Transplantation (HSCT) is the best curative option for many patients with Acute Myeloid Leukemia and related malignancies, mostly due to the antileukemic activity of immune cells contained in the graft. Although in most of the cases HSCT can accomplish apparent disease eradication, frequently residual leukemic cells are able to evade elimination, eventually outgrowing and resulting in clinical relapse. Apparently indistinguishable from disease at diagnosis, relapses commonly display a more aggressive behavior, and most of the available therapeutic options are ineffective. Next-generation sequencing provides the opportunity to track specific alterations and disease subclones during the clinical history of patients, and from this information to identify mechanisms by which leukemic cells evade elimination.
Serial disease samples collected longitudinally during the complex clinical history of a patient with high-risk myelodysplastic syndrome were analyzed by genomic HLA typing and by high-depth exome sequencing (minimum depth of coverage 70x). Patient fibroblasts and mononucleated peripheral blood cells harvested at remission served as reference for germline HLA typing and exome sequence. Whenever possible, leukemic blast were FACS-purified. Sequencing data were aligned to the reference genome using the BWA aligner and analyzed using the tools developed from the Cancer Genome Analysis group at the Broad institute to identify newly acquired mutations, clonal segregation of novel and pre-existing mutations and quantitative clonal dynamics.
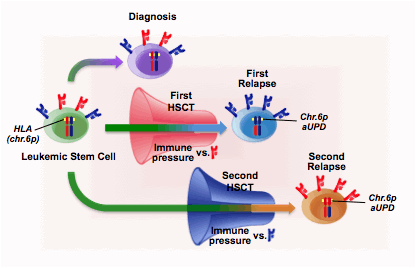
A 54 year old patient with high-risk refractory anemia with excess blasts (pancytopenia, blasts 19%, adverse cytogenetics) underwent unmanipulated T cell-repleted HLA-haploidentical HSCT in the presence of active disease. After one year of remission a first relapse occurred. HLA typing of the leukemic blasts harvested at relapse demonstrated the selective genomic loss of the mismatched HLA haplotype targeted by donor T cells, a frequent mechanism of leukemia immune evasion after haploidentical HSCT, first described by our group (Vago et al, N Engl J Med, 2009). The patient was re-transplanted from a different haploidentical donor mismatched for the HLA alleles retained by the mutated variants; accordingly, T cells from this donor were expectedly alloreactive against the relapsed leukemia. The patient re-obtained complete remission, which was maintained for five years, before the occurrence of a second relapse. Unexpectedly, at this relapse leukemic cells had lost the HLA molecules mismatched with the second donor, and expressed those that had gone amiss at first relapse. Since at both relapses HLA haplotype loss was due to a stable genomic alteration, chromosome 6p acquired uniparental disomy (aUPD), any linear relation between the first and second relapse could be ruled out (Figure 1). Leukemic samples at diagnosis, first relapse and second relapse were further characterized by high-depth exome sequencing, revealing that most of the mutations present in each of the three samples were not found in the other two: only a minute fraction of the mutations could be identified in all three disease presentations, possibly comprising the “driver mutations” harbored by the original leukemic stem cell clone. Amongst these shared mutations we are currently validating the functional and epidemiological relevance of missense alterations in the TP53 tumor suppressor gene, in the NHEJ1 gene (related to chromosomal instability), and in the BTNL8 gene, encoding the costimulatory receptor B7-H5, which may putatively play a role in leukemia recognition by T cells.
Our results demonstrate that antileukemic immunity has a profound impact on leukemia clonal evolution, driving the selection of immuno-privileged subclones. Importantly, the genetic alterations shared between the leukemic variants that emerged at years of distance during the clinical history of our patient suggest the long-term persistence of a reservoir of leukemic stem cells which are resistant to, or in stable equilibrium with, the immune pressure that sculpted their progeny. Finally, the longitudinal approach adopted in this study holds promise for the identification of leukemia “driver” mutations, to provide new insights into leukemogenesis and new rationales for targeted eradicating therapies.
Bordignon:MolMed SpA: Employment. Bonini:MolMed SpA: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal