

Abstract
The frequent occurrence of persistent or relapsed disease after induction chemotherapy in AML necessitates a better understanding of the clonal relationship of AML in various disease phases. In this study, we used SNP 6.0 array-based genomic profiling of acquired copy number aberrations (aCNA) and copy neutral LOH (cnLOH) together with sequence analysis of recurrently mutated genes to characterize paired AML genomes. We analyzed 28 AML sample pairs from patients who achieved complete remission with chemotherapy and subsequently relapsed and 11 sample pairs from patients with persistent disease after induction chemotherapy. Through review of aCNA/cnLOH and gene mutation profiles in informative cases, we demonstrate that relapsed AML invariably represents re-emergence or evolution of a founder clone. Furthermore, all individual aCNA or cnLOH detected at presentation persisted at relapse indicating that this lesion type is proximally involved in AML evolution. Analysis of informative paired persistent AML disease samples uncovered cases with 2 coexisting dominant clones of which at least one was chemotherapy sensitive and one resistant, respectively. These data support the conclusion that incomplete eradication of AML founder clones rather than stochastic emergence of fully unrelated novel clones underlies AML relapse and persistence with direct implications for clinical AML research.
Key Points
These data demonstrate that incomplete eradication of AML founder clones rather than emergence of unrelated novel clones underlies AML relapse.
Introduction
Despite significant advances in the understanding of the biology of adult acute myelogenous leukemia (AML), overall survival remains poor for the more than 12 000 people diagnosed each year in the United States due chiefly to the high rate of relapse after achieving complete remission as well as primary failure of induction chemotherapy.1 Efforts to further unravel the mechanisms leading to relapse and primary refractory disease are critical to guide the development of effective and durable treatment strategies for AML.
Several fundamental questions regarding the underlying causes of disease relapse and persistence in AML remain unanswered. First, although the hierarchy of leukemic progenitors has become increasingly well defined, the specific step within this developmental schema at which a clone achieves the genomic aberrations necessary to emerge or re-emerge as a dominant leukemic clone is unknown.2-6 In addition, the contribution of recently identified recurrently mutated genes to leukemogenesis as well as disease persistence and relapse is only beginning to be understood.7-14 Likewise, the timing of the emergence of acquired copy number aberrations (aCNA) and copy neutral loss-of-heterozygosity (cnLOH), the stability of these genomic lesions in recurrent and refractory AML, and the manner in which they influence disease behavior require further investigation.15-22 From the perspective of treatment of AML, the precise cell compartments in the hierarchy that must be eliminated to affect a cure in vivo have not been identified, and to that end, the relationship of the dominant leukemic clone at diagnosis to that present in relapsed or persistent disease needs to be further elucidated. If the relapsed or persistent clones share a common proximal ancestry with the presenting clone, this implies the inability of conventional chemotherapy to eradicate the founder clone.
With respect to this final point, early studies of recurrent chromosomal abnormalities using conventional cytogenetics in paired presentation and relapsed AML showed that the majority of cytogenetically abnormal cases retained or gained additional abnormalities, whereas a minority lost some or all of their previously identified abnormalities or in rare cases appeared to be unrelated to the diagnosis clone.23-25 Similarly, low resolution SNP array analysis of 27 pairs of diagnosis and relapse adult AML specimens showed that 10 of 11 cases with genomic lesions at diagnosis maintained or added to those lesions at relapse, suggesting proximal clonal relatedness.26 Likewise, whole genome sequencing of 8 AML diagnosis and relapse pairs compared somatic mutation patterns, indicating the resilience of the dominant diagnosis clone or one of its subclones at relapse.27 Recently, whole genome copy number assessment successfully derived the clonal relatedness of diagnosis and relapsed acute lymphoblastic leukemia, and a similar approach may be useful with AML; the addition of longitudinal assessment of recurrently mutated genes will likely augment this approach as well.28
Therefore, to further investigate the relationship of pre and posttreatment dominant and clinically relevant AML clones, we used complementary genomic assessments of clonal architecture using ultra-high resolution SNP 6.0 arrays combined with resequencing of recurrently mutated genes and determination of MLL-PTD status in AML in paired specimens of both relapsed and persistent cases of AML. We demonstrate that some copy number aberrations and gene mutations are acquired early in leukemogenesis and never lost and that the failure to eradicate the leukemic founder clone using currently available conventional therapy underlies relapse and persistence of AML.
Methods
Patients
This study is based on 39 patients with AML for which either paired enrollment or relapse samples or persistent disease samples were available. The patients were enrolled into this study at the University of Michigan Comprehensive Cancer Center. The study was approved by the University of Michigan Institutional Review Board (IRBMED No. 2004-1022) and written informed consent was obtained from all patients before enrollment in accordance with the Declaration of Helsinki. Cytogenetic risk stratification was determined according to Southwest Oncology Group (SWOG) criteria incorporating updated guidelines based on the SWOG AML trial S0106.29
Cell isolation
Ficoll gradient separation and cryopreservation.
Peripheral blood or bone marrow mononuclear cells from AML patients were isolated as described.30
Microbead-based negative selection and subsequent flow cytometry sorting of leukemia specimens.
AML blast DNA used for SNP 6.0 profiling was extracted from negatively column enriched AML cell samples as described30 that were further purified as follows: post-Miltenyi column samples were washed and stained with FITC-conjugated anti-CD33, PE-conjugated anti-CD13, and APC-conjugated anti-CD45 (all antibodies: eBioscience). After final washing, propidium iodide (PI) was added to a concentration of 1 μg/mL to discriminate dead cells. Sorting of cells was done on a FACS-ARIA high-speed flow cytometer (Becton Dickinson). Live cells (PI-negative) were gated for blasts by identifying those cells with intermediate-intensity staining for CD45 and low to moderate-intensity side scatter.31 CD33 and CD13 were then used to further discriminate myeloid blasts versus contaminating erythroid lineage or other nonmyeloid cells. Cells forming a dense population cloud on the CD33 versus CD13 plots that were either single marker positive or double positive were sorted. Aliquots of final cell preparations were made for the majority of sorted samples and manually analyzed for cell composition using cytospins and blasts counts. Results are summarized in supplemental Table 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Preparation of sample DNA
DNA was extracted from highly purified AML blast preparations and paired buccal DNA as described.30
Array data analysis
The DNA was prepared for hybridization to SNP 6.0 arrays according to the manufacturer's recommendations. Affymetrix CEL files for each blast and buccal sample were analyzed using Genotyping Console Version 2.0 (Affymetrix) software for initial quality control, followed by use of the Affymetrix “Birdseed” algorithm to generate tab-delimited SNP call files in text format. Call rates for the entire group of samples included in this report were between 94.02% and 99.35%, with a mean call rate of 98.06%; none of the tumor DNA samples gave out-of-bounds results.
Sample copy number heatmap displays were obtained from CEL files through use of the freely available software dChip,32 adapted to run on a 64-bit computer environment. To generate functional and practical displays of LOH, a 2-step, internally developed, Java-based software analysis system was used. The Pre-LOH Unification Tool (PLUT) served to align all individual patient SNP calls to their respective dbSNP rsID numbers and genomic physical positions before incorporation into the LOH tool version 2, an updated version of the LOH tool able to accommodate Affy SNP 6.0 array data.33
For genomic copy number analysis, 3 observers visually inspected parallel heatmap copy number images of AML blast and paired normal DNA samples generated through dChip and using the median smoothening functionality. Only those copy number changes detected in blast DNA that were not found at the same position in paired normal DNA were called somatic. AML case No. 15 did not have paired normal DNA; therefore, only lesions > 1 Mb (as polymorphic CNVs are rarely > 1 Mb)34 and lesions that were new in the relapse as opposed to the presentation sample were enumerated. Lesions had to be at least 30 SNP positions in length to be scored positive. Using this conservative approach, the shortest identified aCNA lesion was 0.062 Mb in length. The majority of aCNA were defined by > 100 consecutive SNP positions (see supplemental Tables 2-3).
For LOH analysis between paired samples, a filter setting within the LOH tool version 2 was used, allowing visualization of individual paired SNP calls as LOH only if present within 3000 base pairs of another such call. This step filtered out many false, sporadically distributed single LOH calls because of platform noise. Further, LOH calls for at least 3 closely spaced SNPs were required to make an LOH call in any particular genomic region. SNP 6.0 array data files for all patient samples analyzed have been deposited in the GEO public database (accession No. GSE41646).
Genomic losses and gains were also independently nominated using a published algorithmic lesion calling approach.17 This algorithm was developed to be highly specific but slightly less sensitive then visual approaches thus avoiding lesion overcalling that is common with unsupervised algorithmic approaches. Overall, visual and algorithmic approaches demonstrated excellent agreement in lesions called. The discordant calls between visual and algorithmic calling approaches (almost always of the type visual positive and algorithmic negative, see supplemental Table 4) were operationally resolved according to the following rules: (1) if visual loss positive and reconfirmed positive and algorithmic negative and the ratio of the mean copy numbers estimates as determined through dChip for each individual lesion in tumor DNA divided by the mean copy numbers estimates in paired buccal DNA < 0.8 the call is positive, (2) if visual gain positive and reconfirmed positive and algorithmic negative and the ratio of the mean copy numbers estimates for each lesion in tumor DNA divided by the mean copy numbers estimates in paired buccal DNA > 1.33 the call is positive, and (3) if visual gain negative and reconfirmed negative and algorithmic positive and the ratio of the mean copy numbers estimates for each lesion in tumor DNA divided by the mean copy numbers estimates in paired buccal DNA > 1.33 the call is positive.
Methods for algorithmic lesion calling
Median-smoothened copy number data were exported from dChip and were transformed by raising all values to the 0.25 power to approximately stabilize the variance. Then, within each subject, the transformed normal DNA value was subtracted from the transformed tumor DNA value to create a subject-level difference summary. Using this difference summary, we then constructed running average and running variance statistics centered on each SNP, based on uniformly weighted windows of size 30 (for the average) and 120 (for the variance). We next constructed a pseudo Z-score as Z = square root (30) × A/square root (V), where A is the running average and V is the running variance. Initial lesion calls were based on the rule Z <−12 and A <−0.12 (for losses; equivalent to ∼CN estimates of ≤ 1.3) and Z > 16 and A > 0.12 (for gains; equivalent to ∼CN estimates of ≥ 3). These initial calls were then refined by bridging small gaps between consecutive lesion calls. Gaps between consecutive gains or between consecutive losses were bridged if they were up to 1000 SNP positions in length, and the mean of the Z values within the gap was less than − 2 (for a gap between consecutive losses) or greater than 2 (for a gap between consecutive gains); thus only nominal statistical significance is required to bridge small gaps between lesions that had already been detected at high stringency thresholds. The remaining lesion calls were used for subsequent analysis without a lower size limit.
Exon resequencing of selected recurrently mutated genes in AML
Thirteen genes (CEBPA, DNMT3A, IDH1, IDH2, RUNX1, TET2, ASXL1, BCORL1, NPM1, NRAS, KRAS, FLT3, and TP53) known to be recurrently mutated in AML were analyzed for somatically acquired mutations in all 39 AML presentation samples and all 28 relapsed samples using DNA isolated from FACS-sorted blasts that was subsequently Repli-g (Qiagen) amplified. In addition, genes found mutated in individual AML cases from the persistent case collection were subsequently tested for the persistence of the mutation in paired disease samples. Primers to amplify and sequence exon 12 of human NPM1, exons 13-15 and 20 of human FLT3, exons 2-9 of human TP53, exons 2 and 3 of NRAS and KRAS, exon 2 of IDH1 and exon 4 of IDH2, the CEBPA coding exon, all coding exons of DNMT3A, TET2, ASXL1, BCORL1, and RUNX1 and adjacent intronic sequences, were designed using the primer 3 program (http://frodo.wi.mit.edu/primer3/) and sequence information generated using direct sequencing as described.30 Mutations were confirmed to be somatic using unamplified FACS-sorted blast-derived DNA and paired patient buccal DNA as templates.
MLL-PTD Q-PCR
RNA was prepared from ∼ 2 × 105 to 106 ultrapure sorted AML blasts using the Trizol reagent and resuspended in 30 μL DEPC-treated water. Complementary DNA was made from 5 μL of RNA using the Superscript III first strand synthesis kit (Invitrogen) and random priming. Primers to amplify a MLL-PTD spanning exons 9-3 or 10-3 were F: GTCCAGAGCAGAGCAAACAG and R: ACACAGATGGATCTGAGAGG, and a Taqman-based probe (FAM-AGTGGCTCCCCGCCCAAGTA-NFQ) was purchased from Applied Biosystems (MLL probe). Duplicate amplification reactions included primers/probes, Taqman 2x universal PCR master mix, No AmpErase UNG, and 1 μL of cDNA in a 20 μL reaction volume. Two-step PCR was done at an annealing temperature of 54°C for a total of 45 cycles using a Bio-Rad C1000 thermal cycler and a Bio-Rad CFX96 real-time detector system. Normalization of relative copy number estimates for MLL-PTD RNA was done with the Ct values for PGK1 as reference (Δ Ct mean MLL-PTD minus Ct mean PGK1).
Cytogenetic analysis and FISH
Interphase fluorescence in situ hybridization (FISH) analysis for various loci was performed as described35 using BAC probes as detailed in supplemental Table 5. A total of 200 interphase cells were analyzed per patient. Negative controls were prepared from cryopreserved purified AML blast cells without evidence of the respective copy number aberrations by SNP 6.0 array profiling and were run in parallel with each hybridization. Laboratory reference values for background hybridization are added for commonly used probes.
Results
Patient characteristics
Detailed clinical and biologic characteristics of the 28 and 11 AML patients with relapsed or persistent disease, respectively, analyzed in this study are summarized in supplemental Tables 6 and 7. Treatment regimens given including induction and consolidation regimens, where applicable, and response durations are summarized in supplemental Tables 6 and 7. Within the group of samples relapsed from complete remission (relapsed samples), 82% (N = 23) were newly diagnosed and 18% (N = 5) relapsed at trial enrollment. Of the 5 relapsed cases at enrollment, all achieved a second complete remission followed by another relapse. The mean and median remission duration for the paired enrollment-relapse cases was 303 and 271 days, respectively. Of the paired presentation-relapse samples, none carried a TP53 mutation; this is likely because of the fact that few of these patients achieve a CR with conventional chemotherapy.17
Comparative analysis of aCNA, cnLOH, gene mutations, and karyotypes in AML samples procured before and after chemotherapy-induced complete remission (relapsed AML pairs)
We proceeded to catalog all somatically acquired genomic copy number changes in these AML samples as measured through SNP 6.0 array profiling using visual inspection of simultaneous displays of dChip-based copy number estimates (heatmaps) for AML blast and paired buccal DNA. This approach followed our published method for genomic lesion analysis that has been externally validated using FISH,17 which demonstrated 100% concordance between both methods. Of note, SNP array profiling is highly specific and highly sensitive for genomic lesions present in > 25% of the population of cells analyzed.36 This sensitivity is approximately similar to the detection of gene mutations using direct sequencing of PCR products templated on genomic DNA as used in this report. LOH was catalogued using visual inspection of LOH displays generated using the LOH tool Version 2. Data are summarized in supplemental Tables 2 and 3. SNP 6.0 array data were also analyzed using a combination of algorithmic and visual lesion calling methods as detailed in “Methods.” Results from these analyses are summarized in supplemental Tables 8 to 11 supporting the major conclusions drawn. For paired samples with available cryopreserved cells, we also performed FISH-based enumeration of specific genomic lesions to confirm novel aCNA detected at relapse or to verify specific karyotyping results. FISH analyses confirmed all tested acquired SNP 6.0 array-based aCNA in relapsed samples (see supplemental Table 5). FISH analyses also confirmed the absence of acquired SNP 6.0 array-based aCNA (or rarely possible low-level involvement) in paired enrollment samples (see supplemental Table 5). Finally, to provide additional support for our interpretation of SNP 6.0 array results in paired longitudinal samples, we analyzed results from 9 cases of 2 hematologic malignancies (AML and CLL) that were analyzed twice at different times points. Complete concordance between results for such duplicates were detected as detailed in supplemental Table 12 and displayed for some lesions in supplemental Figure 1.
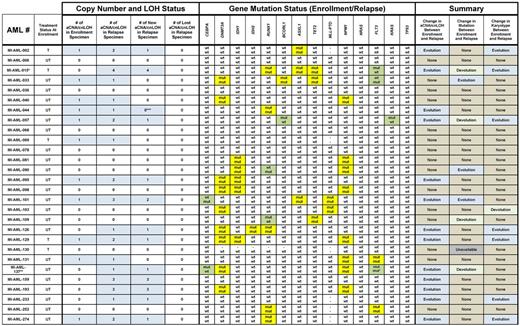
Next, we compared in detail aCNA and cnLOH occurrences, gene mutation patterns and karyotypes for the 28 paired AML samples (presentation − relapse pairs). Data are summarized in Tables 1 and 2 and aCNA heatmaps from representative cases (grouped in triplets; buccal DNA, AML enrollment DNA and AML relapse DNA) are displayed in Figure 1A. Specifically, one of the AML cases (No. 6) carried no informative genomic events. Three additional AML cases (Nos. 36, 68, and 78) carried neither aCNA/cnLOH as detected by SNP-arrays nor gene mutations but harbored chromosomal translocations (typically not detectable using SNP array profiling) that were identical at presentation and at relapse. Seven cases (Nos. 81, 90, 98, 103, 109, 130, and 252) carried no aCNA/cnLOH as detected by SNP-arrays at either presentation or relapse but all these cases at presentation carried at least one gene mutation; all of the gene mutations but 2 were stable (AML case No. 90 gained a RUNX1 mutation at relapse in the setting of retained mutations in IDH1 and NPM1; AML case No. 109 lost a RUNX1 mutation at relapse but retained a TET2 mutation and a t(8;21)(q22;q22) in the majority of cells. Case No. 103 carried a minor subclone at presentation as detected by conventional karyotyping that may have been lost at relapse {changed from 47,XX,dic(8;21)(q21;q22), +dic(8;21)(p21;q22)x2[2]/46,XX[20] to 46,XX[20]} constituting the only case that may have undergone karyotypic devolution. Case No. 103 retained mutations in DNMT3A and MLL at relapse that were present at enrollment).
Summary of aCNA, cnLOH, gene mutations, and karyotype changes in 28 paired AML samples procured at presentation and relapse. Blue fields indicate presence of aCNA or cnLOH or abnormal karyotypes. Yellow fields indicate presence of gene mutations. Green fields indicate a change in the status of a gene mutation. *MI-AML-15 had no germ line DNA available; for this case only CNV > 1 Mb are included in this analysis. **MI-AML-137 shows a different FLT3 ITD between enrollment and relapse. ***The relapse specimen of MI-AML-044 demonstrated elongation of a segment of CN-LOH present in the enrollment specimen.
Summary of karyotypes of 28 paired AML samples procured at presentation and relapse: selected FISH results done for confirmation are presented
| AML no. | Cytogenetics | |
|---|---|---|
| Enrollment karyotype | Relapse karyotype and selected FISH findings | |
| MI-AML-002 | 46,XY,t(9;11)(p22;q23)[9]/45,idem,-Y[7]/46,XY[1] | 45,X,-Y,t(9;11)(p22;q23),del(11)(q22q25)[3]/45,X,-Y,t(2;6;18;4)(q11.2; q22;p11.2;q21),del(9)(q12q22),t(9;11),r(16)(p13;q?22)[3]/46,XX[13] |
| MI-AML-006 | 46,XY[20] | 46,XY[20] |
| MI-AML-015 | 46,XY[20] | 46,XY,del(6)(q13q25)[3]/46,XY[15]; FISH 5% + for del(6)(q13q25) |
| MI-AML-033 | 46,XY,del(3)(q13q27)[16]/46,XY[4] | 46,XY,del(3)(q13q27)[17]/46,XY[2] |
| MI-AML-036 | 46,XY,t(11;19)(q23;p13.1)[20] | 46,XY,t(11;19)(q23;p13.1)[19]/46,XY[1] |
| MI-AML-040 | 46,XY[20] | 46,XY[20] |
| MI-AML-044 | 46,XY,inv(9)(p11q13)c[23] | 45,X,-Y,inv(9)(p11q13)[3]/46,XY,inv(9)(p11q13)[17]; -Y not detected by FISH |
| MI-AML-057 | 46,XY,inv(16)(p13q22)[14] | 46,XY,del(7)(q22q36),inv(16)(p13.1q22)[1]/46,XY[19]; FISH 8.5% + for del(7)(q22q36) |
| MI-AML-068 | 46,XX,t(11;19)(q23;p13.1)[20] | 46,XX,t(11;19)(q23;p13.1)[18]/[46,XY[2] |
| MI-AML-069 | 46,XY,t(6;11)(q21;q23)[1]/47,sl,+der(6)t(6;11)(q21; q23)[1]/46,XY[16] | 46,XY,t(6;11)(q21;q23)[2]/46,XY[18] |
| MI-AML-078 | 46,XX,inv(16)(p13q22)[13]/46,XX[6] | 46,XX,inv(16)(p13q22)[20] |
| MI-AML-081 | 46,XX[20] | 46,XX[20] |
| MI-AML-090 | 46,XX[20] | 46,XX[20] |
| MI-AML-095 | 47,XX,+?4,del(?4)(q2?6q3?2)[2]/46,XX[15] | 47,XX,+?4,del(?4)(q2?6q3?2)[2]/46,XX[18] |
| MI-AML-098 | 46,XY[20] | 46,XY[20] |
| MI-AML-101 | 46,XY[20] | 46,XX[20] |
| MI-AML-103 | 47,XX,dic(8;21)(q21;q22),+dic(8;21)(p21; q22)x2[2]/46,XX[20] | 46,XX[20] |
| MI-AML-109 | 46,XX,t(8;21)(q22;q22)[19]/46,XX[1] | 46,XX,t(8;21)(q22;q22)[12]/46,XX[8] |
| MI-AML-126 | 47,XX,+8[4]/46,XX[16] | 47,XX,+8[3]/47,sl,del(16)(q12q22)[2]/46,XX[15] |
| MI-AML-129 | 47,XX,+8[17]/46,XX[3] | 47,XX,+8[6]/47,sl,del(20)(q11.2q13.3)[6]/47,sl,t(3;9)(q11.2;q22)[4]; FISH 28% + for del(20)(q11.2q13.3) |
| MI-AML-130 | 46,XY[20] | 46,XY[20] |
| MI-AML-131 | 46,XY[20] | 46,XY[20] |
| MI-AML-137 | 46,XY[20] | 46,XY[20] |
| MI-AML-159 | 46,XY,inv(16)(p13q22)[19]/46,XY[1] | 46,XY,inv(16)(p13q22)[19]/46,XY[1] |
| MI-AML-193 | 46,XX[20] | 46,XX[20] |
| MI-AML-233 | 46,XX[20] | 46,XX,t(3;12)(q27;q13)[15]/46,XX[5] |
| MI-AML-252 | 46,XY[20] | 46,XY[20] |
| MI-AML-274 | 45,X,-Y,del(12)(p12p13)[18]/46,XY[2] | 45,X,-Y,del(12)(p12p13)[6]/45,sl,t(6;13)(p12;q12)[7]/46,XY[7] |
| AML no. | Cytogenetics | |
|---|---|---|
| Enrollment karyotype | Relapse karyotype and selected FISH findings | |
| MI-AML-002 | 46,XY,t(9;11)(p22;q23)[9]/45,idem,-Y[7]/46,XY[1] | 45,X,-Y,t(9;11)(p22;q23),del(11)(q22q25)[3]/45,X,-Y,t(2;6;18;4)(q11.2; q22;p11.2;q21),del(9)(q12q22),t(9;11),r(16)(p13;q?22)[3]/46,XX[13] |
| MI-AML-006 | 46,XY[20] | 46,XY[20] |
| MI-AML-015 | 46,XY[20] | 46,XY,del(6)(q13q25)[3]/46,XY[15]; FISH 5% + for del(6)(q13q25) |
| MI-AML-033 | 46,XY,del(3)(q13q27)[16]/46,XY[4] | 46,XY,del(3)(q13q27)[17]/46,XY[2] |
| MI-AML-036 | 46,XY,t(11;19)(q23;p13.1)[20] | 46,XY,t(11;19)(q23;p13.1)[19]/46,XY[1] |
| MI-AML-040 | 46,XY[20] | 46,XY[20] |
| MI-AML-044 | 46,XY,inv(9)(p11q13)c[23] | 45,X,-Y,inv(9)(p11q13)[3]/46,XY,inv(9)(p11q13)[17]; -Y not detected by FISH |
| MI-AML-057 | 46,XY,inv(16)(p13q22)[14] | 46,XY,del(7)(q22q36),inv(16)(p13.1q22)[1]/46,XY[19]; FISH 8.5% + for del(7)(q22q36) |
| MI-AML-068 | 46,XX,t(11;19)(q23;p13.1)[20] | 46,XX,t(11;19)(q23;p13.1)[18]/[46,XY[2] |
| MI-AML-069 | 46,XY,t(6;11)(q21;q23)[1]/47,sl,+der(6)t(6;11)(q21; q23)[1]/46,XY[16] | 46,XY,t(6;11)(q21;q23)[2]/46,XY[18] |
| MI-AML-078 | 46,XX,inv(16)(p13q22)[13]/46,XX[6] | 46,XX,inv(16)(p13q22)[20] |
| MI-AML-081 | 46,XX[20] | 46,XX[20] |
| MI-AML-090 | 46,XX[20] | 46,XX[20] |
| MI-AML-095 | 47,XX,+?4,del(?4)(q2?6q3?2)[2]/46,XX[15] | 47,XX,+?4,del(?4)(q2?6q3?2)[2]/46,XX[18] |
| MI-AML-098 | 46,XY[20] | 46,XY[20] |
| MI-AML-101 | 46,XY[20] | 46,XX[20] |
| MI-AML-103 | 47,XX,dic(8;21)(q21;q22),+dic(8;21)(p21; q22)x2[2]/46,XX[20] | 46,XX[20] |
| MI-AML-109 | 46,XX,t(8;21)(q22;q22)[19]/46,XX[1] | 46,XX,t(8;21)(q22;q22)[12]/46,XX[8] |
| MI-AML-126 | 47,XX,+8[4]/46,XX[16] | 47,XX,+8[3]/47,sl,del(16)(q12q22)[2]/46,XX[15] |
| MI-AML-129 | 47,XX,+8[17]/46,XX[3] | 47,XX,+8[6]/47,sl,del(20)(q11.2q13.3)[6]/47,sl,t(3;9)(q11.2;q22)[4]; FISH 28% + for del(20)(q11.2q13.3) |
| MI-AML-130 | 46,XY[20] | 46,XY[20] |
| MI-AML-131 | 46,XY[20] | 46,XY[20] |
| MI-AML-137 | 46,XY[20] | 46,XY[20] |
| MI-AML-159 | 46,XY,inv(16)(p13q22)[19]/46,XY[1] | 46,XY,inv(16)(p13q22)[19]/46,XY[1] |
| MI-AML-193 | 46,XX[20] | 46,XX[20] |
| MI-AML-233 | 46,XX[20] | 46,XX,t(3;12)(q27;q13)[15]/46,XX[5] |
| MI-AML-252 | 46,XY[20] | 46,XY[20] |
| MI-AML-274 | 45,X,-Y,del(12)(p12p13)[18]/46,XY[2] | 45,X,-Y,del(12)(p12p13)[6]/45,sl,t(6;13)(p12;q12)[7]/46,XY[7] |
![Figure 1. Display of dChip-based heatmap images of representative aCNA in AML. Displayed are heatmaps for lesions organized in triplets [buccal DNA (N), AML blast DNA from enrollment samples (TE) and AML blast DNA from paired relapse (A) or persistent (B) AML samples; TR or TP, respectively]. Genomic losses are indicated by the color blue, genomic gains by the color red.](/view-large/figure/7334211/zh89991201170001.jpeg)
Display of dChip-based heatmap images of representative aCNA in AML. Displayed are heatmaps for lesions organized in triplets [buccal DNA (N), AML blast DNA from enrollment samples (TE) and AML blast DNA from paired relapse (A) or persistent (B) AML samples; TR or TP, respectively]. Genomic losses are indicated by the color blue, genomic gains by the color red.
Display of dChip-based heatmap images of representative aCNA in AML. Displayed are heatmaps for lesions organized in triplets [buccal DNA (N), AML blast DNA from enrollment samples (TE) and AML blast DNA from paired relapse (A) or persistent (B) AML samples; TR or TP, respectively]. Genomic losses are indicated by the color blue, genomic gains by the color red.
Eleven cases were characterized by presence of variable numbers of aCNA/cnLOH at presentation. Of these, 55% (6/11) of cases gained additional aCNA/cnLOH, one case acquired a FLT3-ITD (No. 33) and case No. 44 elongated a pre-existing cnLOH lesion. Six additional cases without aCNA/cnLOH at presentation gained aCNA/cnLOH at relapse and 3 of 6 of these demonstrated karyotypic evolutions (Table 2). Therefore, acquisition of novel aCNA/cnLOH at relapse was a common event occurring in 46% (13/28) of these cases.
Importantly, we detected no case that demonstrated loss at relapse of any of the aCNA/cnLOH that was detected at presentation.
In contrast to the aCNA/cnLOH/karyotype analysis detailed in the preceding paragraphs, review of the gene mutation pattern of 14 commonly mutated genes analyzed here was more variable and demonstrated 2 patterns: (1) absolute stability of the mutation (DNMT3A, IDH1, IDH2, ASXL1, TET2 and NPM1) or (2) stability of the mutation with occasional loss or gain in individual cases (detected for CEBPA, RUNX1, BCORL1, FLT3-ITD and KRAS).
The combined multidimensional review of aCNA, cnLOH, karyotypes, and gene mutations in paired presentation-relapse AML samples, allowed us to derive various scenarios for clonal relatedness in these samples. These are schematically depicted in Figure 2A-C, with TE indicating the measured clonal genomic aberrations in bulk presentation samples and TR indicating the measured clonal genomic aberrations in bulk relapsed samples (AC indicates the inferred genomic changes that existed in an antecedent leukemia clone). Importantly, the combined data from all informative samples together support existence of a chemotherapy-resistant antecedent clone (AC) as the cellular source of AML relapse.

Schema of clonal relatedness in AML cases (paired enrollment and relapse samples) inferred from genomic profiling data. The quantitatively dominant clone as detected at the time of analysis is circled in green. AC: inferred antecedent clone. TE: enrollment sample, TR: paired relapse sample. Arrows indicate possible routes of clonal evolution. Circles indicate gene mutations, rectangles indicate aCNA or cnLOH.
Schema of clonal relatedness in AML cases (paired enrollment and relapse samples) inferred from genomic profiling data. The quantitatively dominant clone as detected at the time of analysis is circled in green. AC: inferred antecedent clone. TE: enrollment sample, TR: paired relapse sample. Arrows indicate possible routes of clonal evolution. Circles indicate gene mutations, rectangles indicate aCNA or cnLOH.
Comparative analysis of aCNA, cnLOH, and gene mutations in AML samples procured before and after chemotherapy (persistent disease pairs)
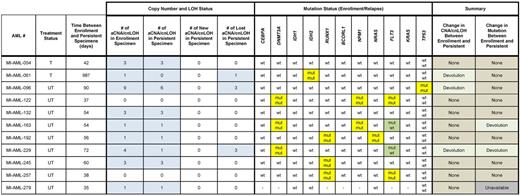
Within the group of AML cases that displayed overt resistance to potent induction chemotherapy as judged by hematopathologic analysis of interim routine clinical staging bone marrows, the following patterns for aCNA, cnLOH, and gene mutations emerged (see Table 3): 2 cases (Nos. 122 and 257) without aCNA/cnLOH before or after chemotherapy carried stable gene mutations. Five additional cases with aCNA/cnLOH at presentation carried the same genomic lesions and gene mutations before and after chemotherapy. These data for 7 AML cases together indicate presence at enrollment of a dominant chemorefractory AML clone. Three cases (Nos. 61, 96, and 229) with distinct aCNA/cnLOH present at enrollment lost some but not all of these aCNA/cnLOH and gained none after initial induction therapy; 1 additional case (No. 163) lost a FLT3-ITD. Together, data from these 4 cases indicate loss of at least one initially codominant chemosensitive subclone and persistence of at least one codominant chemoresistant subclone. The aCNA heatmap data from representative cases (grouped in triplets; buccal DNA, AML enrollment DNA and AML persistent DNA) are displayed in Figure 1B. Results are also schematically depicted in Figure 3A and B, with TE indicating the measured clonal genomic aberrations in bulk presentation samples and TP indicating the measured clonal genomic aberrations in bulk persistent disease samples (AC indicates the inferred genomic changes that existed in an antecedent leukemia clone).
Summary of aCNA, cnLOH and gene mutations in 11 AML samples at presentation and at disease persistence following chemotherapy. Blue fields indicate presence of aCNA or cnLOH or abnormal karyotypes. Yellow fields indicate presence of gene mutations. Green fields indicate a change in the status of a gene mutation.

Schema of clonal relatedness in AML cases (paired enrollment and persistent disease samples) inferred from genomic profiling data. The quantitatively dominant clone as detected at the time of analysis is circled in green. AC: inferred antecedent clone. TE: enrollment sample, TP: paired persistent disease sample. Arrows indicate possible routes of clonal evolution. Circles indicate gene mutations, rectangles indicate aCNA or cnLOH.
Schema of clonal relatedness in AML cases (paired enrollment and persistent disease samples) inferred from genomic profiling data. The quantitatively dominant clone as detected at the time of analysis is circled in green. AC: inferred antecedent clone. TE: enrollment sample, TP: paired persistent disease sample. Arrows indicate possible routes of clonal evolution. Circles indicate gene mutations, rectangles indicate aCNA or cnLOH.
Discussion
In this study, we used ultra-high density SNP 6.0 array-based genomic profiling together with sequence analysis of 13 commonly mutated genes and MLL-PTD assessment to interrogate the genomes of highly purified blasts from AML patients before and after treatment with chemotherapy. These data constitute the first study in AML that incorporates measurements of aCNA and cnLOH, recently identified novel gene mutations and karyotypes into one comprehensive genomic analysis, therefore extending results derived from either low density SNP arrays, mutation profiling of selected genes or genome-wide next gen sequence analysis of a limited number of AML sample pairs. The study is furthermore based on DNA isolated from FACS-sorted blasts, which removes confounding effects of AML and DNA purity on data interpretation. Through comparison of patterns of aCNA, cnLOH, karyotypes, and gene mutations in paired samples the following conclusions are supported by this data: (1) genomic aCNA/cnLOH are involved early in AML pathogenesis/evolution, (2) persistence in complete clinical remission of dominant antecedent and chemotherapy-refractory clones underlied AML relapse in all informative cases in this study, (3) evidence for stochastic emergence of clones at relapse that are completely unrelated to the antecedent clones was not found, and (4) AML disease persistence (primary refractory disease) after potent chemotherapy either represents persistence of a dominant chemorefractory clone or reflects persistence of chemorefractory clone(s) and loss or substantial numerical reduction of a coexisting chemosensitive clone(s).
AML pathogenesis is driven by multiple recurrent genomic events. These include aCNA and cnLOH (for instance del5q, del7q, cnLOH for FLT3-ITDs and many others), chromosomal translocations, gene mutations, and epigenetic deregulations.37-39 Although the recurrent nature of these genomic aberration categories indicates importance to AML pathogenesis, the exact contribution of each of these genomic aberrations to AML evolution, response to chemotherapy and AML relapse or persistence are still incompletely understood. Although some gene mutations (for instance NPM1, IDH1, IDH2, DNMT3A, ASXL1, TET2, MLL-PTD) are stable in paired AML analysis, others are not (for instance CEBPA, FLT3, RUNX1, BCORL1, or K-RAS), indicating a hierarchical and qualitatively different contribution to the AML phenotype.13,40-46 Data in this report support that aCNA/cnLOH that are detected in AML blasts at disease presentation are not lost at relapse, indicating a proximal and fundamental/driver role in AML pathogenesis; this finding is in contrast to a hypothetical model in which all aCNA/cnLOH in AML are late-stage, and at times stochastic, genomic acquisitions and is in contrast to findings derived from paired analysis of selected gene mutations in pediatric AML which indicated frequent changes.
The data presented here, together with recently published data based on deep sequencing of 8 paired AML genomes, clearly demonstrate that incomplete eradication of dominant antecedent clones underlies AML relapse and persistence.27 This scenario is partly different from what has been described in a subset of ALL using similar approaches in which emergence of clones that evolutionarily predated and that were distinct from the dominant clones at disease presentation became dominant at relapse.28 The scenario in AML also is different from a hypothetical scenario in which a “fertile cellular soil” or a “predisposed aberrant bone marrow” would allow for frequent and stochastic emergence of novel unrelated AML clones at relapse after eradication of the antecedent clone that initially became the clinically apparent and symptoms-causing clone.
Given incomplete eradication of dominant antecedent AML clones after current therapy approaches in AML, the question arises what cell type contributes to the phenotypical resistance and to the clinical relapse in patients? A separate question centers on the molecular and cellular determinants of drug resistance in cases with wild-type TP53. Both of these questions are not addressed by these data. Are all critical genomic aberrations already present in cells that give rise to and are able to generate the bulk of the leukemia blasts (which ultimately constitute clinically apparent leukemia) or do rare leukemia blasts that harbor all critical genomic aberrations survive chemotherapy and constitute the source of relapse in humans? Clearly, in the setting of persistent refractory disease, drug resistant AML blasts survive and if one were to consider AML relapse a consequence of persistence of smaller amounts of resistant blasts then such blasts and cells that can generate more blasts need to be targeted by additional and better therapies early on to achieve disease control.
Given that founder clones are the source of relapse in AML, one of the more immediate applications of these findings relates to the routine identification of the persistence of such founder clones in postinduction/consolidation disease remission states. To improve such monitoring, which is akin to MRD analyses, one would have to incorporate multiple genomic tests in parallel and focus on gene mutations or aCNA that are never lost at relapse. One may also apply biologic assays of AML generation, such as xenografts, to remission marrows to detect occult leukemia. Therapeutically, given the current lack of approved noncross-resistant therapeutics, the early use of allogeneic transplantation for patients with high risk of relapse remains the most promising approach. Long-term, the identification of therapies post induction that can eliminate the founder clones is essential and may be aided by screens in AML xenografts.
In summary, these data based on complementary genomic technologies (SNP array profiling and gene resequencing) that are targeted to the detection of dominant clones (the clones that cause clinically overt AML) rather than rare subclones, suggest that incomplete eradication of antecedent founder clones, as opposed to emergence of new and unrelated clones, underlies AML relapse and persistence and that targeting such clones more effectively may improve outcome in AML.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful for services provided by the microarray core of the University of Michigan Comprehensive Cancer Center.
This work was supported by the Scholars in Clinical Research Program of the Leukemia & Lymphoma Society of America (S.M.). This research is supported (in part) by the National Institutes of Health through the University of Michigan's Cancer Center Support Grant (5 P30 CA46592) and Oncology Research Training Grant (T32 CA 009357-30).
National Institutes of Health
Authorship
Contribution: B.P., P.O., Y.L., and S.N.M. performed the laboratory research; B.P. and S.N.M. analyzed clinical data; K.S. and C. Li assisted with statistical analysis and software development for data analysis; J.K., C. Lam, and D.R. performed the FISH and karyotype analyses; S.N.M. conceived the study and supervised the work; and B.P., P.O., and S.N.M. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sami N. Malek, Dept of Internal Medicine, Division of Hematology and Oncology, University of Michigan, 1500 E Medical Center Dr, Ann Arbor, MI 48109-0936; e-mail: smalek@med.umich.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal