

Abstract
Abstract 2594
Abnormalities affecting long arm of chromosome 3 are rare but recurrent in Acute Myeloid Leukemia (AML) and are detected in a variable percentage of AML patients according to different series. The 2008 World Health Organization classification recognizes AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2) as a distinct subtype characterized by a poor prognosis. Allogeneic stem cell transplantation seems to improve outcome in eligible patients with these aberrations. Inappropriate expression of the ecotropic viral integration site 1 (EVI1) was demonstrated in virtually all patients with t(3;3)(q21;q26.2) and inv(3)(q21q26.2); as well as in a majority of patients with other 3q26 rearrangements. Other chromosome 3 abnormalities are rarely recognized in AML patients; clinical and prognostic relevance of these alterations is not yet defined. The aim of this study is to assess the prognostic impact of chromosome 3 abnormalities on disease characteristics and treatment outcome in AML.
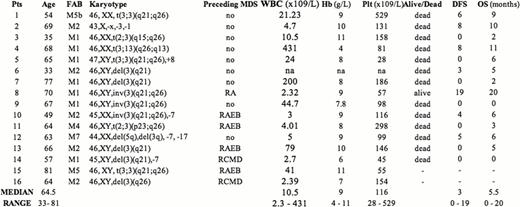
A total of 580 consecutive adult patients were diagnosed with AML at our institution between February 2002 and July 2012. Conventional cytogenetic analysis performed on diagnostic bone marrow samples detected the presence of 3q abnormalities in 16 patients (2.7%). Two patients were lost to follow-up and were not evaluated for survival analysis. Molecular status of FLT3 and NPM1 was also performed and results are available for 10 patients. Median follow-up time for patients in this series was 47 months ( range 6–125).
There were 10 male and 6 female patients, the median age being 64.5 years (range 33–81), 10 patients had de novo AML while 6 evolved from a previously diagnosed myelodysplastic syndrome (MDS). Karyotype from MDS phase was available in 2 patients; both acquired 3q rearrangement at time of progression to AML. At time of diagnosis median haemoglobin value was 9.0 g/dL (range 4–11); median leucocyte count was 10.5 × 103̂/L (range 2.3 – 431). Median platelet count was 116 × 109̂/L (range 28 – 529), consistently with previous studies, which have shown that these patients present with higher platelet count at diagnosis when compared with no 3q rearranged ones. Regarding cytogenetic features 3 patients had t(3;3)(q21;q26), 3 patients had inv(3) (q21; q26), 3 patients showed a balanced rearrangement involving 3q26, while 6 patients harbored a del3q. One patient showed monosomy 3. Additional chromosomal changes were demonstrated in 5 patients, two of them had a complex karyotype (see Table 1), 3 had a monosomy 7. Thirteen patients out of 14 received intensive induction chemotherapy; complete remission (CR) was achieved in 5 patients (CR rate: 35.7%), the remaining 7 patients were resistant to induction as well as to salvage chemotherapy. Four patients underwent autologous stem cell transplantation. Median overall survival in this series is 5.5 months (range 0 – 20). At present only one patient is still alive and in CR, 20 months after diagnosis. Median disease free survival (DFS) for patients achieving a CR was 9 months (range 6–20). Median overall survival for patients resistant to first-line therapy was 3 months (range 0–6). Clinical features and treatment outcome of the patients are summarized in Table 1.
The incidence of 3q abnormalities in our single institution series is 2.4%, in keeping with previous studies. Our findings confirm the association between these alterations and thrombocytosis at diagnosis, preceding MDS or multilineage dysplasia, presence in all FAB subtypes (except M3), association with additional chromosomal abnormalities as well as the poor response to conventional chemotherapy (CR rate 35.7%), and very short DFS in spite of obtaining CR.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal