

Abstract
Abstract 466
The genetic lesions identified to date do not fully recapitulate the molecular pathogenesis of chronic lymphocytic leukemia (CLL) and do not entirely explain the development of severe complications, such as chemorefractoriness. BIRC3, along with TRAF2 and TRAF3, cooperate in the same protein complex that negatively regulates MAP3K14, the central activator of non-canonical NF-kB signaling. Following the initial observation of recurrent BIRC3 mutations in splenic marginal zone lymphomas, we performed targeted re-sequencing of the BIRC3 coding sequence and splicing sites across the spectrum of B-cell neoplasia. By this screening, BIRC3 mutations were identified only in CLL (2/20), while were absent in diffuse large B-cell lymphoma (0/30), Burkitt lymphoma (0/38), follicular lymphoma (0/20), extranodal marginal zone lymphoma (0/65), hairy cell leukemia (0/19), and multiple myeloma (0/22). This observation prompted the investigation of prevalence and clinical impact of BIRC3 genetic lesion in CLL. The study population included 4 clinical CLL panels representative of different disease phases: i) a cohort of fludarabine-refractory CLL (n=49); ii) a cohort of fludarabine-sensitive CLL (n=62); iii) clonally related Richter syndrome (RS) (n=33; all diffuse large B cell lymphomas); and iv) a consecutive series of newly diagnosed and previously untreated CLL (n=308). BIRC3 was analyzed for mutations by Sanger sequencing (exons 2–9) and for copy number abnormalities by FISH (probe RP11-177O8). BIRC3 was affected in 12/49 (24%) fludarabine-refractory CLL by inactivating mutations (7 frameshift and 1 non-sense) and gene deletions (n=7) that distributed in a mutually exclusive fashion with TP53 disruption (p=.004) (Fig 1A and 1B). Two fludarabine-refractory CLL carried a biallelic inactivation of the BIRC3 gene by mutation of one allele and deletion of the second allele. All inactivating mutations were somatically acquired, were predicted to generate aberrant transcripts carrying premature stop codons, and caused elimination or truncation of the C-terminal RING domain, whose E3 ubiquitin ligase activity is essential for MAP3K14 proteosomal degradation by BIRC3. Western blot analysis confirmed the expression of aberrant bands corresponding in size to the predicted truncated BIRC3, and revealed constitutive non-canonical NF-kB activation in fludarabine-refractory CLL harboring BIRC3 disruption by mutations or deletion, as documented by NFkB2 processing from p100 to p52 (Fig. 1). BIRC3 was the sole gene of the TRAF3/MAP3K14-TRAF2/BIRC3 complex targeted by molecular lesions after extensive mutation and FISH analysis of TRAF2, TRAF3 and MAP3K14 in the fludarabine-refractory CLL cohort (n=49). To investigate whether BIRC3 genetic lesions were restricted to chemorefractory cases, we analyzed BIRC3 for mutations and deletions in other CLL subsets. Fludarabine-sensitive CLL were consistently devoid of BIRC3 disruption in all cases (n=62), suggesting that BIRC3 genetic lesions specifically associate with a chemorefractory phenotype among CLL requiring treatment (Fig. 1A). BIRC3 disruption was restricted to 1/33 (3.0%) clonally-related RS, corroborating the notion that RS is molecularly distinct from chemorefractory progression without transformation (Fig. 1A). In a consecutive series evaluated at CLL diagnosis, BIRC3 disruption was rare (13/308, 4%, all TP53 wild type), associated with primary chemorefractoriness among cases requiring treatment, and predicted poor outcome (Fig. 1A). By univariate analysis, the crude impact of BIRC3 disruption on survival was a ∼5.3 fold increase in the hazard of death (HR: 5.31; 95% CI: 2.62–10.73) and a significant overall survival (OS) shortening (median OS in BIRC3 disrupted cases: 3.1 years vs not reached in BIRC3 wild type cases; p<.001) (Fig. 1D). Multivariate analysis selected BIRC3 disruption as an independent risk factor of OS (HR: 4.25; 95% CI: 1.74–10.36; p=.001) after controlling for confounding clinical (age and Rai stage) and genetic (IGHV mutation status, 11q22-q23 deletion, TP53 disruption, NOTCH1 mutations) variables at diagnosis. These data document that disruption of BIRC3, a critical regulator of non-canonical NF-kB signaling, associates with fludarabine-refractoriness among TP53 wild type CLL and identifies a subgroup of patients showing poor outcome.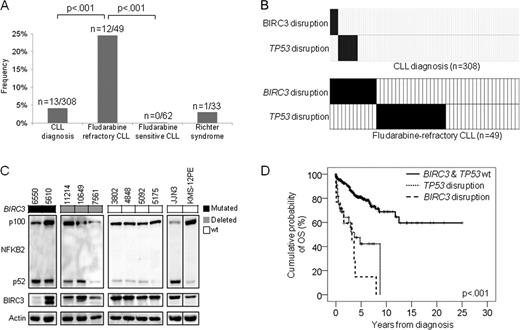

Disclosures:
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal